Glucocorticoids
Evangelia Charmandari
Tomoshige Kino
George P. Chrousos
Introduction
Glucocorticoids regulate a broad spectrum of physiologic functions essential for life and play an important role in the maintenance of basal and stress-related homeostasis (1,2,3). Approximately 20% of the genes expressed in human leukocytes are regulated positively or negatively by glucocorticoids (4). Glucocorticoids are involved in almost every cellular, molecular, and physiologic network of the organism and play a pivotal role in critical biologic processes, such as growth, reproduction, intermediary metabolism, immune and inflammatory reactions, as well as central nervous system and cardiovascular functions (1,2,3,4). Physiologic amounts of glucocorticoids are also essential for normal renal tubular function and thus for water and electrolyte homeostasis. Furthermore, glucocorticoids represent one of the most widely used therapeutic compounds often employed in the treatment of inflammatory, autoimmune, and lymphoproliferative disorders (1,2,3).
The adrenal glands consist of the adrenal cortex and the adrenal medulla. In the fetus, the adrenal cortex is divided into two zones, the outer definitive zone and the inner fetal zone. The latter represents approximately 80% of the adrenal volume and secretes weak androgens with the delta-5 configuration (dehydroepiandrosterone, DHEA). After birth, the fetal zone regresses rapidly during the first 2 weeks and disappears almost completely by the third month of life. A few islands of cells remain which may later develop into androgen-secreting cells. During infancy and childhood, the definitive zone and perhaps remnants of the fetal zone evolve into the adult adrenal cortex, which consists of three anatomic zones: the outer zona glomerulosa, the intermediate zona fasciculata, and the inner zona reticularis. The zona glomerulosa is responsible for the production of aldosterone, the zona fasciculata for the production of cortisol, and the zona reticularis for the production of adrenal androgens. The adrenal medulla is functionally related to the sympathetic nervous system and secretes epinephrine and norepinephrine both at basal conditions and in response to stress. The adrenal cortex and medulla are embryologically and functionally interdependent, influencing each other’s organogenesis, growth and function (1,2).
Pathways of Steroid Biosynthesis
The synthesis of the glucocorticoid cortisol and the mineralocorticoid aldosterone are under the control of regulatory systems that largely function independently. The major regulation of the former is by the adrenocorticotropic hormone (ACTH), while the latter is regulated primarily by the renin-angiotensin system and circulating potassium ions. All steroid hormones produced by the adrenal cortex are derived from cholesterol. Low-density lipoprotein (LDL) cholesterol is the major source of cholesterol utilized in adrenal steroidogenesis; however, the adrenal cortices can synthesize cholesterol de novo. Proteolytic and lipolytic enzymes act on LDL to release cholesterol esters for storage in lipid droplets in the adrenal steroidogenic cells (1,2,3).
In order for the adrenal cortex to synthesize active steroid hormones, a number of changes are required in the structure of cholesterol. Several of these reactions are catalyzed by the steroid hydroxylases, which are members of a superfamily of genes known collectively as cytochromes P450 (CYP). Adrenal steroidogenesis follows three distinct routes, which reflect the zonal differences in terms of function and regulation (Fig. 51.1).
The rate-limiting step in steroid biosynthesis is importation of cholesterol from cellular stores to the matrix side of the mitochondrial inner membrane, where the cholesterol side chain cleavage system is located. This import is controlled by the steroidogenic acute regulatory (StAR) protein, the synthesis of which is increased by trophic adrenocortical stimuli, such as ACTH (3,5,6). In addition to StAR protein, cholesterol transfer is also mediated by another protein, the peripheral benzodiazepine receptor (7) (Fig. 51.2). The first enzymatic step in steroid biosynthesis common to all steroidogenic pathways takes place in the mitochondrion and leads to the cleavage of six carbon atoms from the side chain of cholesterol, converting this C27 compound to the C21 steroid pregnenolone. This reaction is known as cholesterol side-chain cleavage and is catalyzed by the cytochrome P450 enzyme CYP11 A (P450 scc, cholesterol desmolase, side-chain cleavage enzyme), which is an integral protein of the inner mitochondrial membrane (1,2,3,8). Pregnenolone, the common precursor for
all steroids, then passes by diffusion from the mitochondrion to the cytoplasm where it undergoes further metabolism by several other enzymes (Fig. 51.2).
all steroids, then passes by diffusion from the mitochondrion to the cytoplasm where it undergoes further metabolism by several other enzymes (Fig. 51.2).
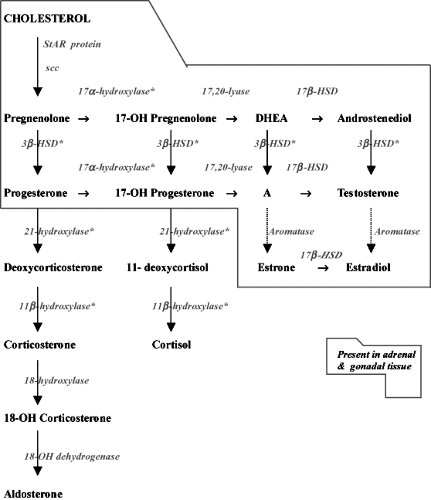 Figure 51.1. Schematic representation of adrenal steroidogenesis. Solid lines: major pathway. Dotted lines: major pathway in ovaries and testes but minor in the adrenals. *: deficient enzymatic activity results in CAH. A, androstenedione; DHEA, dehydroepiandrosterone; 3β-HSD, 3β-hydroxysteroid dehydrogenase; 17β-HSD, 17β-hydroxysteroid dehydrogenase; scc, cholesterol side-chain cleavage enzyme; StAR: steroidogenic acute regulatory protein. |
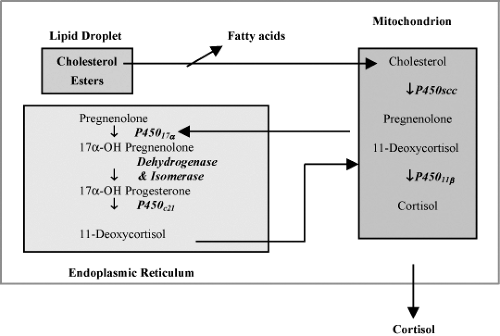 Figure 51.2. Pathway of biosynthesis of cortisol in the adrenal cortex. (Reproduced from Simpson ER, Waterman MR. Steroid biosynthesis in the adrenal cortex and its regulation by adrenocorticotropin. In: DeGroote LJ, Besser M, Burger HG, et al., eds. Endocrinology, Philadelphia, PA: W.B. Saunders, 1995:1630–1641.) |
To synthesize mineralocorticoids in the zona glomerulosa, the microsomal 3β-hydroxysteroid dehydrogenase (3β-HSD) converts pregnenolone to progesterone (3,9). The latter is 21-hydroxylated in the cytoplasm by CYP21 (P450c21, 21-hydroxylase) to produce deoxycorticosterone (DOC). Aldosterone, the most potent 17-deoxysteroid mineralocorticoid compound, is produced by 11β-hydroxylation of DOC to corticosterone, followed by 18-hydroxylation and 18-oxidation of corticosterone (Fig. 51.1). The final three steps in aldosterone synthesis are accomplished by a single mitochondrial P450 enzyme, CYP11 B2 (P450aldo, aldosterone synthase) (10).
To produce cortisol, CYP17 (P450c17, 17α-hydroxylase/17,20-lyase) in the endoplasmic reticulum of the zona fasciculata and zona reticularis converts pregnenolone to 17α-hydroxypregnenolone (3,11). 3β-HSD in the zona fasciculata utilizes 17α-hydroxypregnenolone as a substrate, producing 17α-hydroxyprogesterone. The latter is 21-hydroxylated by CYP21 to form 11-deoxycortisol, which is further converted to cortisol by CYP11B1 (P450c11, 11β-hydroxylase) in the mitochondria (Fig. 51.2).
In the zona reticularis of the adrenal cortex and in the gonads, the 17,20-lyase activity of CYP17 converts 17α-hydroxypregnenolone to DHEA, a C19 steroid and sex steroid precursor. DHEA is further converted by 3β-HSD to androstenedione. In the gonads, androstenedione is reduced by 17β-hydroxysteroid dehydrogenase (12). In pubertal ovaries, aromatase (CYP19, P450c19) can convert androstenedione and testosterone to estrone and estradiol, respectively (13). Testosterone may be further metabolized to dihydrotestosterone by steroid 5α-reductase in androgen target tissues (14).
The enzymatic differences between the zona glomerulosa and the zona fasciculata can be summarized by stating that the zona glomerulosa lacks P450c17α but has the ability to catalyze the 18-oxidation of corticosterone, the precursor of aldosterone. Interestingly, this activity has been shown to be a property of the 11β-hydroxylase, which appears to be capable of 18-hydroxylation and subsequent reduction to the aldehyde aldosterone.
Regulation of Cortisol Secretion
Plasma glucocorticoid concentrations are regulated in ways that reflect the varying physiologic needs for the hormones under basal conditions and in response to stress. Cortisol secretion is primarily regulated by the ACTH, a 39-amino acid peptide released from the anterior pituitary in response to the hypothalamic neuropeptides corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP). CRH is the strongest and probably the most important stimulator of ACTH secretion. AVP also activates secretion of ACTH and potentiates the effect of CRH on ACTH secretion. Catecholamines, angiotensin II, oxytocin, atrial natriuretic peptide, cholecystokinin, vasoactive intestinal polypeptide, and pituitary adenylate cyclase-activating polypeptide may also contribute to the secretion of ACTH (1,2,3).
ACTH is synthesized as part of a large precursor molecule of 214 amino acid, proopiomelanocortin. Depending on the cleavage enzymes, which are expressed in a tissue-specific fashion, ACTH and several other peptides, for example, NH2-terminal peptide, joining peptide, β- or #979;-lipotropins, β-endorphin, α-melanocyte-stimulating hormone (αMSH), and corticotropin-like intermediate peptide, are produced. The first 18 amino acids of the N-terminus of ACTH are conserved through species and carry its biological activity. Thus, synthetic ACTH (1 to 24) and ACTH (1 to 18) are often used for clinical purposes instead of the full-length ACTH. The sequence of αMSH, which is contained within the ACTH molecule, stimulates the production of melanin by the melanocytes, causing skin and mucosal hyperpigmentation when secreted in excess (1,2,3).
ACTH is secreted in regular pulses of variable amplitude over 24 hours, with peak concentrations attained in the early morning hours (04:00 AM to 08:00 AM), thus forming the basis of the circadian pattern of ACTH and cortisol secretion (1,2,3,15). The acute action of ACTH is to increase the flux of cholesterol through the steroidogenic pathway, resulting in rapid production of steroids. ACTH also influences the remaining steps of steroidogenesis, as well as the uptake of cholesterol from plasma lipoproteins, thus ensuring a continuous supply of cholesterol to the mitochondria to meet the demands of activated pregnenolone biosynthesis. It also maintains the size of the adrenal glands.
CRH is the principal hypothalamic factor that stimulates the pituitary production of ACTH, while AVP plays a synergistic role (16). Both neuropeptides are produced by parvocellular neurons of the paraventricular nuclei of the hypothalamus, but are also found in other parts of the central nervous system, as well as in peripheral non-CNS locations (17). CRH and AVP are secreted in a pulsatile fashion that results in the episodic secretion of ACTH and the circadian variation of cortisol secretion. The frequency of the hypothalamic pulsations of CRH and AVP is relatively constant, while the amplitude of these secretory episodes increases in the early morning hours and during stress, resulting in major changes in the plasma concentrations of cortisol. Therefore, it is the magnitude of each pulse of CRH/AVP and consequently ACTH that determines the total daily cortisol secretion. In addition to ACTH, other factors may also play an important role in the regulation of the adrenal cortex, including neural factors, medullary peptides, and immune system products (18,19).
Cortisol is the primary negative regulator of basal hypothalamic–pituitary–adrenal (HPA) axis activity through negative feedback upon the pituitary, the hypothalamus and suprahypothalamic centers, such as the hippocampus (20). Thus, both ACTH and CRH secretion are inhibited by glucocorticoids. This effect is mediated by the classic glucocorticoid receptor α (GRα) in the pituitary and hypothalamus and by both GRα and the mineralocorticoid receptor (MR) in the hippocampus. The MR is in fact functioning as a GR, granted that the enzyme 11β-dehydrogenase type 2, which converts cortisol to the inactive cortisone, is not present in this tissue to protect it from the mineralocorticoid effects of cortisol. The concentrations of aldosterone are too low to compete with cortisol for the hippocampal MRs. Whether and to what extent direct glucocorticoid feedback
on the adrenal cortex itself regulates cortisol synthesis is not clear (21).
on the adrenal cortex itself regulates cortisol synthesis is not clear (21).
Secretion and Metabolism
In normal subjects, the secretion of glucocorticoids follows a diurnal pattern, with peak concentrations observed between 06:00 AM and 08:00 AM and lowest concentrations around midnight (22). The cortisol production rate is approximately 10 to 15 mg per m2 per day (23). More than 90% of circulating cortisol, and to a lesser extent aldosterone, is bound tightly to corticosteroid-binding globulin (CBG) or transcortin (24). The remaining (10%) of the circulating cortisol is free or loosely bound to albumin. The free and albumin-bound fractions of cortisol represent the biologically active form of the hormone. When plasma cortisol concentrations exceed 20 μg per dL, CBG becomes fully saturated and most of the excess cortisol is biologically active. CBG is synthesized in the liver. Estrogens, thyroid hormones, pregnancy, and oral contraceptives are associated with increased CBG concentrations (24,25,26), whereas hypercortisolism, hepatic, or renal disease result in decreased CBG concentrations. In the presence of an intact HPA axis, alterations in CBG concentrations are likely not to affect circulating free cortisol concentrations.
The primary site of cortisol metabolism in humans is the liver, kidney, and a number of target tissues. Cytosolic and microsomal enzymes, including cytochrome P450, 5α/5β-reductase, 3α/3β-oxidoreductase, and 11β-hydroxysteroid dehydrogenase (11β-HSD), play an important role in the hepatic metabolism of cortisol (27,28,29). The major routes of hepatic metabolism involve A-ring and side-chain reduction followed by conjugation with glucuronic acid and sulphate (30). The inactive glucuronide and sulphate metabolites are excreted by the kidneys, whereas only less than 1% of cortisol is excreted unchanged in the urine. Therefore, the metabolic clearance of cortisol is influenced primarily by factors altering hepatic clearance and to a significant degree by renal enzymes and factors affecting renal excretion.
11β-hydroxysteroid dehydrogenase (11β-HSD) is known to modulate glucocorticoid action in tissues by altering the rate of conversion between cortisol and cortisone. There are two types of 11β-HSD: 11β-HSD type 1 (11β-HSD1) and type 2 (11β-HSD2). 11β-HSD1 acts as a reductase, catalyzing the conversion of inactive cortisone to active cortisol, and may contribute to the tissue hypersensitivity to glucocorticoids. 11β-HSD1 is widely expressed in the liver, lung, adipose tissue, vascular components, ovary, and the CNS. Increased expression of 11β-HSD1 activity in adipose tissue is associated with visceral obesity, indicating that 11β-HSD1 can enhance glucocorticoid action in local tissues (29).
In contradistinction to 11β-HSD1, the physiologic importance of the other 11β-HSD2 is well documented. Patients harboring mutations in this enzyme develop symptoms of mineralocorticoid excess, while mice bearing a deletion in this gene demonstrate glucocorticoid-dependent mineralocorticoid excess. 11β-HSD2 acts as a dehydrogenase that converts cortisol to inactive cortisone. 11β-HSD2 is expressed in classic aldosterone-sensitive tissues, such as the kidney, colon, sweat glands, and the placenta, where it protects the MR from the mineralocorticoid effects of cortisol, allowing the much lower aldosterone concentrations to exert such actions (29).
Molecular Mechanisms of Glucocorticoid Action
At the cellular level, the actions of glucocorticoids are mediated by an intracellular receptor protein, the GR, which functions as a hormone-activated transcription factor that regulates the expression of glucocorticoid target genes (1,31,32). The human (h) GR belongs to the superfamily of steroid/thyroid/retinoic acid receptor proteins (31). The hGR gene consists of 9 exons and is composed of a poorly conserved amino-terminal domain (NTD); a central, highly conserved DNA-binding domain (DBD); and a carboxyl-terminal, ligand-binding domain (LBD) (1,31,32). The NTD contains a major transactivation domain, termed activation function (AF)-1, which is located at amino acids 77 to 262 of the hGR. The DBD spans over amino acids 420 to 480 and contains two zinc finger motifs through which the receptor binds to specific DNA sequences in the promoter region of target genes, the glucocorticoid-response elements (GREs). The LBD contains a second transactivation domain, AF-2, as well as sequences important for interaction with heat shock proteins (HSPs), nuclear translocation, and receptor dimerization (33,34,35) (Fig. 51.3A,B).
Alternative slicing of the hGR gene in exon 9 generates two highly homologous receptor isoforms, termed α and β. These are identical through amino acid 727, but then diverge, with hGRα having an additional 50 amino acids and hGRβ having an additional, nonhomologous 15 amino acids (36) (Fig. 51.3A). hGRα is ubiquitously expressed in almost all human tissues and cells, resides primarily in the cytoplasm, and represents the classic GR that functions as ligand-dependent transcription factor. hGRβ is also ubiquitously expressed in tissues usually at lower concentrations than hGRα, with the exception of epithelial cells and neutrophils (36). In contradistinction to hGRα, hGRβ resides primarily in the nucleus of cells independently of the presence of ligand, does not bind glucocorticoids, and exerts a dominant negative effect upon the wild-type hGRα (32,37).
In the absence of ligand, hGRα resides primarily in the cytoplasm of cells as part of a large multiprotein complex, which consists of the receptor polypeptide, two molecules of hsp90, and several other proteins (38). The hsp90 molecules are thought to sequester hGRα in the cytoplasm of cells by maintaining the receptor in a conformation that masks or inactivates its nuclear localization signals (NLSs). Upon hormone binding, the receptor undergoes conformational changes, which result in dissociation from hsp90 and other proteins, unmasking of the NLSs and exposure of the ligand-binding pocket. In its new conformation, the activated, ligand-bound hGRα translocates into the nucleus, where it binds as homodimer to GREs located in the promoter region of target genes. hGRα then communicates with the basal transcription machinery and regulates the expression of glucocorticoid-responsive genes positively or negatively, depending on the GRE sequence and promoter context (38) (Fig. 51.4). The receptor can also modulate gene expression, as a monomer, independently of GRE binding, by physically interacting with other transcription
factors, such as activating protein-1, nuclear factor-κB, and signal transducers and activators of transcription (38).
factors, such as activating protein-1, nuclear factor-κB, and signal transducers and activators of transcription (38).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


