Genetics & Metabolism
Alyssa R. Letourneau
Marsha F. Browning
Vivian E. Shih
Inborn Errors of Metabolism
Definition
Inherited enzyme mutation alters metabolism → excess or lack of certain metabolites
Incidence
1:1400–200,000 live births; NBS screens for many of the disorders
Neonatal Presentation
(Pediatrics 1998;102:E69; Vademecum Metabolicum 2004:3)
24–72 hr of life
Ill infant w/ nonspecific sx’s: Lethargy, diff feeding, vomiting, abn resp, hypotonia and szr’s
Abnormal body or urinary odor
MSUD: Urine smells of maple syrup or burnt sugar
Isovaleric acidemia and glutaric acidemia type II → pungent, unpleasant “sweaty feet” odor
Late Presentation
(Crit Care Clin 2005;21:S9; Vademecum metabolicum 2004:3)
>28 d of life; recurrent vomiting, lethargy, or fasting → coma with nonfocal neuro exam
Liver dysfxn + mental status changes
Adolescent/Adults Presentation
(J Inherit Metab Dis 2007;30:631)
Psychiatric d/o, recurrent rhabdo, myoglobinuria, cardiomyopathy
Acute recurring confusion → urea cycle defect, porphyria, homocysteine remethylation defect
Chronic psych sx’s → homocystinuria, Wilson dz, adrenoleukodystrophy, some lysosomal d/o
Mild mental retardation and personality Δ → homocystinuria, nonketotic hyperglycemia, other.
Specific Triggers of Decompensation
(Vademecum Metabolicum 2004:3)
Vomiting, fasting, infection, fever, vaccinations, surgery, accident or injury, changes in diet → protein or carbohydrate metabolism disorders
High- protein diet and/or catabolic state → aminoacidopathies, organic acidemia, urea cycle def
Fruit, sugar (sucrose), liquid medicines → fructose intolerance
Lactose → galactosemia
High fat → Fatty acid oxidation disorders
Drugs → porphyria, glucose-6-phosphate-dehydrogenase deficiency
Extensive exercise → disorders of fatty acid oxidation, glycolysis, respiratory chain
Emergencies
Critical Labs
(Pediatrics 1998;102:E69; Vademecum Metabolicum 2004:4)
Stat D-stick, CBC w/diff, chem 7, blood gas, NH3, lactate, plasma and urine amino acids, U/A, urine reducing substances, urine ketones, urine organic acids, ESR
CRP, CK, ALT, AST, coagulation studies
Store plasma samples for amino acids, acylcarnitine and filter paper (“Guthrie” card)
If LP done → freeze CSF for further studies
Consider ECG, echo, head imaging
Emergent Treatment
(Pediatrics 1998;102:E69; Vademecum Metabolicum 2004:5)
Start Rx before confirmed dx; stop protein, fat, galactose, and fructose intake
Consult metabolic specialist
1st goal: Remove metabolites (organic acid intermediates or ammonia)
Hyperammonia: Immediate HD for coma, vent dependency, or signs of cerebral edema.
Urea cycle defects: 6 cc/kg of 10% arginine HCL IV over 90 min.
Organic acidemia: Vit B12 (1 mg) IM for B12-responsive form of methylmalonic acidemia. Biotin (10 mg) PO or NGT for biotin-responsive carboxylase deficiency
Acidosis: Give bicarb with frequent ABGs; severely acidotic: HD
2nd goal: Prevent catabolism
IV glucose (calories and substitute for nml liver glucose prod) → D10, 150 cc/kg/d w/ lytes
Stop protein as above, IV lipids for urea cycle defects
If unclear diagnosis: Continue glucose drip, review history
Follow lytes, glucose, lactate, ABG, keep Na >135 to avoid cerebral edema
Ref: www.childrenshospital.org/newenglandconsortium/NBS/Emergency_Protocols.html
Metabolic Acidosis and Ketosis
Definition
(Pediatrics 1998;102:E69; Vademecum Metabolicum 2004:11)
ABG with anion gap ≥16. Decreased pH, HCO3, Paco2 (if resp compensation)
Ketosis present as response to fasting, catabolic state, or ketogenic diet
Presentation
Acute metab encephalopathy → lethargy, poor feeding, vomiting, coma, szr’s, ↓ tone, resp distress, or apnea
Ddx
Renal bicarb loss → renal Fanconi syndrome, RTA, cystinosis, osteopetrosis → tyrosinemia, fructose intolerance, glycogen storage disease type I, mitochondrial diseases, methylmalonic aciduria (chronic renal damage)
GI loss of bicarb → diarrhea
↑ organic acids → lactate production or ketosis →infxn, sepsis, ↑ catabolic state, tissue hypoxia, dehydration, intoxication, DKA
Diagnostic Studies and Management
Labs as above, Rx according to primary diagnosis
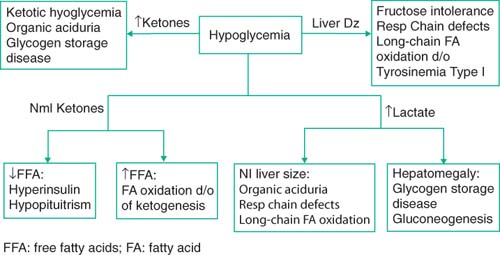
Hypoglycemia
Definition
(Pediatrics 1998;192:E69; Vandemecum Metabolicum 2004:6)
Glucose <2.6 mmol/L (45 mg/dL) at all ages
History and Clinical Manifestations
Determine time since last meal, drugs
Check hepatomegaly, liver failure signs (palmar erythema, spider angiomata, gynecomastia, jaundice), small genitals, hyperpigmentation, short stature
Ddx
Disorders of protein intolerance, carbohydrate metabolism, or fatty acid oxidation
Hepatic glycogen storage dz (except Pompe); no glycogenolysis (worse with fasting)
In neonate: Need to rule out sepsis, SGA, maternal diabetes; maybe slow adaptation
Persistent neonatal hypoglycemia → hyperinsulinemia or hypopituitarism
Labs While Hypoglycemic
As above + insulin, cortisol, lactate, free fatty acids, 3-hydroxybutyrate, Ketostix (urine)
Acylcarnitine (dried blood spots or plasma); for fatty acid ox disorders + organic acidurias, C-peptide level
Spare tube for additional labs
Organic acids in the urine
 |
Treatment
IV glucose at 7–10 mg/kg/min, for calories and to replace normal liver glucose production → D10% glucose, 110–150 cc/kg/d with electrolytes
Keep FS >100; If glucose needs >10 mg/kg/min → likely hyperinsulinism
Hyperammonemia
Definition
(Pediatrics 1998;192:E69; Vandemecum Metabolicum 2004:8)
Suspect metabolic dz in neonate if NH3 >200 μmol/L; All other ages NH3 >100 μmol/L
Ddx
Always consider medication effect
Urea cycle defects (no acidosis) and organic acidemias (+ metabolic acidosis), liver failure
Neonates can have transient hyperammonemia of the newborn → sx’s w/in 24 hr of birth; large premature infants with pulm dz. Usually no recurrent hyperammonemia
Labs
Labs as above; must have uncuffed venous or arterial sample, on ice, sent stat
AA in plasma and urine, organic acids and orotic acid in urine
Acylcarnitine in dried blood spots
Treatment
Must contact metabolic team immediately
NH3 >500 → central line, art line, HD
1st infusion: 12 cc/kg D10 over 2 hr with lytes
Arginine hydrochloride 360 mg/kg
Na-benzoate 250 mg/kg (alternate pathway for nitrogen excretion)
Carnitine 100 mg/kg
Ondansetron 0.15 mg/kg IV bolus in noncomatose (to prevent N/V)
Follow glucose, add insulin if needed, check ammonia after 2 hr
Ref: Neonate: www.childrenshospital.org/newenglandconsortium/NBS/neonate2.html
Infant/child: www.childrenshospital.org/newenglandconsortium/NBS/infant_child.html
Abnormal Newborn Screen
Newborn Screen
(Vademecum Metabolicum 2004:53)
www.childrenshospital.org/newenglandconsortium/NBS/Emergency_Protocols.html
Started in the 1960s for PKU → broadened to many disorders w/ regional variations
Galactosemia
Definition
(Pediatrics 1998;102:E69; Pediatrics 2006;118:E934; Vademecum Metabolicum 2004:3)
Lactose is broken down into glucose and galactose for absorption
AR deficiency in enzymes → accumulation of glactose-1-phosphate and galactitol
Incidence
“Classic galactosemia”: Most common with galactose-1-phosphate uridyltransferase (GALT) deficiency → 1:47,000 newborns
Galactokinase (GALK) def → 1:1,000,000; Galactose-4′-epimerase (GALE) def → rare
Presentation
Progressive jaundice and liver dysfunction
First 2 wk of life → V/D, poor weight gain, cataracts, indirect hyperbili from hemolysis
Most states screen for it on NBS:
Galactose (total) 20–30 mg/dL; No Rx. Outpatient review
Repeat screening of dried blood spot, galactose, GALT activity
Galactose (total) 30–40 mg/dL; repeat as above
Start lactose-free milk, outpt review, follow-up repeat labs
Galactose (total) >40 mg/dL; hospitalization, lactose-free diet
Check liver and kidney function, coagulation and ultrasound
Check galactose, galactose-1-phosphate, GALT activity
Treatment
Galactose-free formula
If ill: Supportive care, Vit K, FFP, Abx for presumed Gram-negative sepsis and phototherapy for hyperbilirubinemia
Usually improve w/ removal of galactose; check all meds after dx (many contain galactose)
Prognosis
Persistent liver disease, cataracts, mental retardation
Many die of E. coli sepsis
Phenylketonuria/Phenylalaninemia
Definition
(Pediatrics 2006;118:E934; Vademecum Metabolicum 2004:71)
Abnormal increase in amino acid phenylalanine in blood
Deficiency of liver enzyme phenylalanine hydroxylase → impairs neurotransmitter production
If >20 mg/dL with accumulation of phenyl ketones → phenylketonuria (PKU)
Can be a benign process
Incidence
1:13,500–19,000 for PKU; non-PKU hyperphenylalaninemia 1:48,000
AR w/ >400 mutations; if untreated → severe brain damage, MR, seizures, spasticity
Treatment
Early Rx important → admit to the hospital, inverse relationship between time to treatment and IQ
Positive NBS → check quantitative level of phenylalanine and tyrosine concentration
Provide with medical protein sources low in phenylalanine
Follow phenylalanine levels and keep low
Management of Known Inborn Errors of Metabolism
Urea Cycle Disorders
Definition
(Vademecum Metabolicum 2004)
Inherited enzyme and transport protein def w/ ↓ removal excess NH3 from protein metabolism
Incidence
Most common inborn errors of metabolism; 1:8000
Usually presents after newborn period, at all ages
Presentation
Neonates → lethargy, poor feeding, hyperventilation, seizures, encephalopathy
Infants/children → FTT, feeding problems, vomiting, neuro sx’s, lethargy, ataxia, szr’s
Teens/adults: Chronic neuro or psych sx’s, behav probs, disorientation, lethargy, psychosis
Diagnosis
Based on abnormal plasma and urine amino acid levels
Ornithine Transcarbamylase Deficiency (OTC)
Most common; 1:14,000 incidence; X-linked, many w/ residual enzyme activity
Can present between 1 mo of age to childhood with significant illness; inc urine orotic acid
Treatment
Acute therapy → see previous discussion
Long-term management: Metabolic team to assess diet, low protein diets, good fluid intake, vaccinations, and treating infections early.
Aminoacidopathies
Definition
(Vademecum Metabolicum 2004:57)
Def of enzymes for AA metab → toxic substances accumulate in brain, liver, and kidneys
If known disorder of AA metabolism and ill → call metabolic team
Labs as above and emergency Rx glucose as above, stop protein intake, keep Na >140 to prevent cerebral edema, Abx, detox prn w/ diuresis or HD, Vits and carnitine depending on dz
Tyrosinemia
(Am J Med Genet C Semin Med Genet 2006;142C:121: Vademecum Metabolicum 2004:72)
Accumulation of tyrosine in fluids and tissue
Type I is the most severe → liver failure, neurologic crises, rickets, hepatocarcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


