Genetics in Pediatric Primary Care
Tyler Reimschisel
Ada Hamosh
Many health care professionals consider genetic diseases to be limited to single-gene disorders, such as sickle cell disease, cystic fibrosis (CF), muscular dystrophy (MD), and phenylketonuria. But genes play a much larger role in human disease and health. For example, McCandless et al. have shown that up to 71% of children admitted to a children’s hospital have a disorder with a significant genetic component. In fact, all human traits and diseases, including common conditions such as otitis media, are modified by multiple genetic factors. Furthermore, variation in the genetic background of individuals with a given disease or syndrome explains in part the variable clinical manifestations of that disease or syndrome. For example, generalists are well aware of variation in common medical problems such as asthma, attention deficit-hyperactivity disorder, and streptococcal pharyngitis. Just as the environment can influence the severity of these medical problems, host and microbial genes also play a role in the clinical presentation. As the Human Genome Project draws to completion and the entire genetic code of humans is deciphered, we will learn much more about how genes play a role in disease and health. This chapter reviews the current understanding of genetics and its role in the medical problems that the pediatrician may encounter.
GENETIC TERMINOLOGY AND INHERITANCE PATTERNS
A basic understanding of genetic terminology is essential for comprehending genetic principles. A gene is the basic unit of genetic information. It is composed of a sequence of nucleotides in DNA within chromosomes. A codon is a triplet of three nucleotides or bases, and each codon encodes a single amino acid in a polypeptide. A gene may contain many exons and introns. Exons are sequences of DNA that are transcribed and processed into mature messenger RNA (mRNA), which is the template for translating nucleotide information into polypeptides. Introns also are nucleotide sequences of DNA that are transcribed into RNA, but the intronic sequences are spliced out to make mRNA during post-transcriptional processing. Thus, mature mRNA is composed only of RNA that is complementary to the DNA sequences of the exons of a particular gene.
Variability can exist in the DNA sequence at a particular genetic locus. These variants are known as alleles. Most genes have a single DNA sequence that is known as the wild-type allele. All permanent changes to the DNA sequence of the wild-type allele are known as mutations, and alleles that contain mutations are called mutated or variant alleles. A mutated sequence of DNA that occurs in 1% or more of the population is known as a polymorphism. Less common alleles are known as rare variants. If a mutation in a gene causes a disease, then the mutation is a pathogenic or deleterious mutation. Frequently, the term “mutation” is used to denote a change in the DNA that causes disease, but the term pathogenic mutation is preferable. In an individual, the DNA sequence in a gene is the genotype of that individual. The phenotype in that individual is the biochemical, molecular, and clinical manifestations of the genotype. The genome is the entire genetic information of an individual. Genome also can refer to the entire genetic information of a population or species.
Many genetic diseases manifest genetic heterogeneity, in which more than one pathogenic mutation can lead to the same disease. For example, CF can be caused by more than 1,000 different pathogenic mutations in the CF transmembrane conductance regulator (CFTR) gene. This is an example of a specific type of genetic heterogeneity known as allelic heterogeneity, in which different mutations at the same locus or gene lead to the same disease. Another example of genetic heterogeneity is locus heterogeneity, in which mutations in different genes lead to the same disease. Locus heterogeneity occurs in a variety of genetic diseases, including Ehlers-Danlos syndrome, tuberous sclerosis, and alpha- and beta-thalassemias.
Alternatively, pathogenic mutations in the same gene can cause distinct clinical phenotypes. This is termed phenotypic heterogeneity. For example, many mutations in CFTR cause CF, but some mutations in CFTR cause congenital bilateral absence of the vas deferens without a predilection for pulmonary disease. One of the most striking examples of phenotypic heterogeneity is that different mutations in the gene lamin A/C cause seven distinct diseases, including progeria (premature aging syndrome), a type of dilated cardiomyopathy, early-onset atrial fibrillation, a type of Emery-Dreifuss MD, limb-girdle MD type 1B, Charcot-Marie-Tooth disease type 2B, familial lipodystrophy, mandibuloacral dysplasia, and lipoatrophy with diabetes, hepatic steatosis, hypertrophic cardiomyopathy, and leukomelanodermic papules.
Phenotypic heterogeneity is distinct from pleiotropy, another feature that distinguishes many genetic diseases from other etiologies of disease. Pleiotropy is the manifestation of multiple, seemingly unrelated, medical problems due to a single gene abnormality. For example, dysfunction of the chloride channel in CF causes recurrent pulmonary infections, pancreatic insufficiency, and infertility in males. Smith-Lemli-Opitz syndrome, a disorder of cholesterol synthesis, causes mental retardation, two- to three-toe syndactyly and other dysmorphic features, urogenital abnormalities, and a low 7-dehydrocholesterol level.
In individuals with a given disease, each feature of the phenotype can present with varying degrees of severity or can be completely absent. This variation in the phenotype of a single disease, termed variable expressivity, can occur in individuals with a disease who are from different families and in affected individuals within the same family. The genetic factors that cause the variable expressions of a disease include allelic heterogeneity, locus heterogeneity, and differences in the genes that modify the function and expression of the abnormal gene product. An individual’s unique phenotype also is related to his or her
environmental exposures and stochastic or chance molecular and biochemical events that occur in the individual.
environmental exposures and stochastic or chance molecular and biochemical events that occur in the individual.
Variable expression of a phenotype should not be confused with penetrance. Penetrance is the statistical likelihood that individuals with a particular genotype will have any phenotypic expression, no matter how mild or severe. If a mutation in a gene does not always cause a phenotypic expression, then that gene shows reduced penetrance. For example, in some families retinoblastoma does not develop in up to 40% of individuals with a pathogenic mutation in the retinoblastoma susceptibility gene. Unlike expressivity, penetrance is either “full” or “reduced” by a particular percentage; it should not be called “variable.” Some diseases show reduced or incomplete penetrance. All diseases have variable expression in their phenotype.
Types of Genetic Alterations
During DNA replication, multiple errors can occur. However, at least 99.9% of these errors are corrected. The efficiency of repairing DNA replication errors ensures the stable transfer of genetic information during multiple generations of cell division. It also minimizes the frequency of genetic diseases due to single-gene defects. Yet, not all errors are corrected, and one error occurs for every 10-10 base pairs per cell division. Thus, less than one mutation occurs per cell division.
Some mutations only change a single coding nucleotide base. These point mutations can have different molecular effects. For example, a missense mutation causes a change in the amino acid that is encoded by that codon. A nonsense mutation introduces a base pair that makes the codon a stop codon. The introduction of a premature stop codon causes mRNA degradation in a process called nonsense mediated decay (NMD). Other point mutations may disrupt RNA processing by changing a splice site that defines exon–intron boundaries. These mutations lead to frameshift mutations and NMD, or abnormal polypeptide formation. For example, sickle cell disease occurs because a single base substitution (adenine → thymine) at codon 6 changes the sixth amino acid from glutamic acid to valine in the gene that encodes the beta-chain of hemoglobin. This single change causes deoxygenated hemoglobin to polymerize and “sickle.”
Some genes normally contain a series of three repeating base pairs or trinucleotides. Pathogenic mutations can occur if a trinucleotide sequence in a gene increases in number during gametogenesis. For example, fragile X syndrome occurs when the trinucleotide cytosine-guanine-guanine in the FMR1 gene increases from the normal repeat length of 6 to 45 repeats to more than 200 repeats. The length of these trinucleotide repeats also can decrease. Thus, expanded trinucleotide repeats are called dynamic mutations. Frequently, the repeating trinucleotide encodes a series of glutamines. The current hypothesis for the pathogenesis of many trinucleotide repeat diseases is that polyglutamine aggregates cause abnormal protein folding and function or are toxic to the cells. Because the trinucleotide repeat is a dynamic mutation, trinucleotide repeat diseases manifest anticipation, in which the disease can present at a younger age and with a more severe phenotype as the mutation expands in size from one generation to the next. Trinucleotide repeat diseases include Huntington disease, most autosomal dominant spinocerebellar ataxias, Friedreich ataxia, and myotonic dystrophy.
In addition to point mutations, small intragenic deletions, duplications, and insertions can occur. If the number of bases in a deletion, duplication, or insertion is a multiple of three, then there will be a loss or gain of amino acids in the translated protein. However, if the number of bases is not a multiple of three, then the mutation causes a frameshift in the DNA sequence that usually leads to mRNA decay because a stop codon occurs prematurely.
Large deletions, duplications, insertions, and inversions also can occur. They can affect many genes and noncoding regions within a chromosome and lead to an abnormal phenotype. Some of these abnormalities can be identified through the microscopic analysis of a high-resolution karyotype. Others are too small to see by light microscopy. These submicroscopic abnormalities can be identified by fluorescent in situ hybridization (FISH).
Deletions, duplications, insertions, and inversions can alter the number of bases in the DNA sequence. An insertion and deletion can occur if a translocation occurs of a piece of one chromosome into the sequence of a different chromosome. If the translocation occurs without an obvious loss of genetic information on karyotype analysis, then it is a balanced translocation. If it appears on the karyotype that genetic material has been lost, then it is an unbalanced translocation. Both balanced and unbalanced translocations can cause an abnormal phenotype, because the translocation can disrupt multiple gene sequences, even if no obvious loss of genetic material is present on the karyotype. A translocation can be balanced in a parent and become unbalanced in the offspring. Loci with balanced translocations are prone to errors during mitosis and meiosis because of independent segregation of sister chromatids. An inversion is a mutation in which the orientation of the DNA sequence is reversed. Once again, depending on the size and location of the inversion, this mutation may present as an abnormal phenotype in the individual or the individual’s offspring.
Aneuploidy is another chromosomal alteration in which a change occurs in the number of chromosomes. For example, trisomy 21 or Down syndrome is due to an extra copy of chromosome 21. Turner syndrome is due to the loss of an X chromosome in females (45, X).
Inheritance Patterns
Single-gene disorders can be inherited in a variety of Mendelian patterns, including autosomal dominant, autosomal recessive, and X-linked (Fig. 11.1). A dominantly inherited disease occurs when only one of the two alleles at a particular gene locus has a pathogenic mutation. A recessively inherited disease occurs when both alleles have pathogenic mutations. When one allele has a pathogenic mutation and the other allele has the wild-type base pair sequence, the affected individual is said to be heterozygous at that gene locus. If the identical mutation occurs on both alleles, the affected individual is homozygous at that locus. In recessively inherited diseases, it is common to have different pathogenic mutations on each allele. In those cases, the affected individual is compound heterozygous at that locus. If the gene that has pathogenic mutation(s) is located on one of the 22 autosomes, an autosomal dominant or recessive disorder results. In some cases, a disorder can be inherited in an autosomal dominant pattern in one family and in an autosomal recessive pattern in a different family (e.g., osteogenesis imperfecta type 3). This occurs because some mutations are pathogenic if they change only one allele, whereas other mutations are less disruptive to the gene product and must be present in both alleles to cause a particular disease. If the gene that has the pathogenic mutation is located on the X chromosome, the disease is transmitted in an X-linked pattern.
Autosomal Dominant Inheritance
In disorders that are inherited in an autosomal dominant pattern, males and females are affected equally. The disease can
be inherited from only one parent. Thus, vertical transmission of a disease can be identified in the pedigree of a family with an autosomal dominant condition. Autosomal dominant conditions can also occur sporadically when a gene spontaneously mutates in the gamete of an unaffected parent, and the disease presents for the first time in the offspring. Alternatively, some autosomal dominant disorders “skip generations” when the phenotype is expressed minimally or is not expressed at all (nonpenetrant) in an individual who has the pathogenic mutation in one of his or her alleles. Offspring of an affected individual have a 50% chance of inheriting the mutation. Unaffected individuals do not pass the disease to their offspring unless the disease is nonpenetrant. Transmission of a disease from a father to a son helps to distinguish autosomal dominant conditions from X-linked disorders (discussed in the section, X-linked Inheritance).
be inherited from only one parent. Thus, vertical transmission of a disease can be identified in the pedigree of a family with an autosomal dominant condition. Autosomal dominant conditions can also occur sporadically when a gene spontaneously mutates in the gamete of an unaffected parent, and the disease presents for the first time in the offspring. Alternatively, some autosomal dominant disorders “skip generations” when the phenotype is expressed minimally or is not expressed at all (nonpenetrant) in an individual who has the pathogenic mutation in one of his or her alleles. Offspring of an affected individual have a 50% chance of inheriting the mutation. Unaffected individuals do not pass the disease to their offspring unless the disease is nonpenetrant. Transmission of a disease from a father to a son helps to distinguish autosomal dominant conditions from X-linked disorders (discussed in the section, X-linked Inheritance).
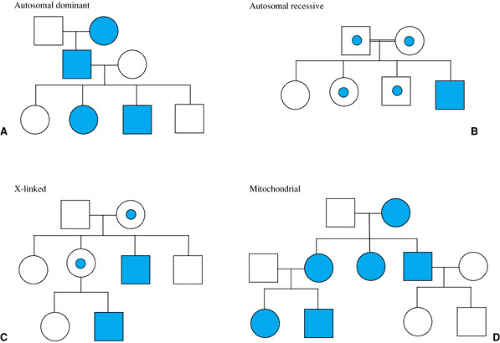 FIGURE 11.1. Typical pedigrees of Mendelian and mitochondrial inheritance patterns. Squares are males; circles are females. Shaded symbols are affected; unshaded are unaffected. Dot in symbol is carrier in autosomal recessive pedigree and obligate heterozygote in X-linked pedigree. Double line denotes consanguinity in the autosomal recessive pedigree. |
At least three molecular pathophysiologic mechanisms exist for autosomal dominant conditions, including dominant loss of function or haploinsufficiency, dominant gain of function, and dominant negative effect. Dominant loss of function or haploinsufficiency occurs when only 50% of the gene product from the wild-type allele has not enough residual function to prevent disease. This can occur when the gene product is a transcription factor that regulates gene expression or when the product is a structural protein (as in some mutations that cause Marfan syndrome). Dominant gain of function occurs when the pathogenic mutation increases the function of the normal gene product (e.g., achondroplasia) or makes the gene product a toxin to the cell (as in some spinocerebellar ataxia syndromes). A dominant negative effect occurs when the pathogenic mutation encodes for an abnormal protein that interferes with the function of the normal gene product (as in some mutations that cause osteogenesis imperfecta and Marfan syndrome).
Autosomal Recessive Inheritance
In autosomal recessive disorders, the parents are usually each carriers of a mutated allele. As carriers, they are asymptomatic and do not manifest any phenotype of the disease. Each parent has a 50% chance of passing the mutated allele to each offspring, and each offspring has a 25% chance of inheriting both mutated alleles and manifesting the phenotype. Consequently, analysis of a family pedigree may reveal multiple full siblings with a specific disease, but the disease does not affect first-degree relatives (parents and offspring), first cousins, or individuals from other generations unless two affected parents have children. In autosomal recessive diseases, males and females are affected equally. There may be a history of consanguinity in the parents of an affected child, especially if pathogenic mutations in a particular gene are rare in the population. When parents have one child with a given autosomal recessive disease, the risk of those parents having another child with the same disease is 25% for each and every pregnancy. Unaffected siblings of an affected proband have a two-thirds risk of being a carrier of the pathogenic mutation.
Many autosomal recessive diseases are inborn errors of metabolism secondary to defective enzyme function, and almost all inborn errors of metabolism are inherited in an autosomal recessive pattern. The carriers of pathogenic mutations in genes that encode enzymes do not usually manifest the disease phenotype, because most biochemical reactions require less than 25% of the normal enzyme activity. Carriers have 50% of the normal enzyme activity, well above the threshold
for dysfunction. Affected individuals develop disease because both alleles carry pathogenic mutations, and the enzyme activity is usually close to 0. Some individuals with a particular metabolic disease may have a milder phenotype. In those cases, the pathogenic mutations encode a protein with some residual enzyme activity, and/or modifier genes in affected individuals minimize the dysfunction of the abnormal gene product.
for dysfunction. Affected individuals develop disease because both alleles carry pathogenic mutations, and the enzyme activity is usually close to 0. Some individuals with a particular metabolic disease may have a milder phenotype. In those cases, the pathogenic mutations encode a protein with some residual enzyme activity, and/or modifier genes in affected individuals minimize the dysfunction of the abnormal gene product.
X-linked Inheritance
If the affected gene is located on the X chromosome, the disorder that results is inherited in an X-linked pattern. Although medical literature frequently refers to disorders that are “X-linked recessive” or “X-linked dominant,” these terms are not appropriate when discussing X-linked disorders or traits because the X-chromosomes in females undergo lyonization, and males have only one X chromosome (hemizygous). Lyonization is the mechanism whereby one of the two X chromosomes in females is inactivated. It is a random process that occurs in early embryonic development. At that stage, either the paternal or maternal X chromosome is inactivated in each cell. Disorders that are X-linked can show extreme variation of expression in females, based on the percentage of active X chromosomes that carry a pathogenic mutation. If the X chromosome with the pathogenic mutation is preferentially inactivated, then the inactivation is skewed, and the individual has a favorable lyonization pattern for that disorder. In those cases, the female may be asymptomatic or show only mild manifestations of the disease. Alternatively, the X chromosome that is not mutated may be preferentially inactivated. This unfavorable lyonization will lead to varying degrees of clinical manifestations of the disease. Due to variation in X inactivation, many X-linked disorders have variable expression in females, including color blindness, X-linked mental retardation syndromes, and Duchenne MD.
Males have only one X chromosome. They are said to be hemizygous for most genes on the X chromosome; therefore, lyonization does not occur in males. Consequently, any pathogenic mutation on the X chromosome of a male will cause disease. Lyonization in females explains why X-linked diseases in males are more severe than they are in females. Unless an affected female has a pathogenic mutation in the same allele on both X chromosomes, lyonization will usually make some of the X chromosomes without the pathogenic mutation active. The normal allele on this X chromosome will provide a measure of normal gene product that will help diminish the severity of the disease in females. Some X-linked disorders only are manifested in females or are extremely rare in males. These disorders may be lethal in males. Incontinentia pigmenti is one example. However, many X-linked disorders are rare in males because the mutation occurs on the paternal X during gametogenesis. The father passes only the Y chromosome to males (Thomas hypothesis).
Another reason to avoid using the terms “X-linked recessive” and “X-linked dominant” is that female “carriers” of X-linked disorders are not necessarily asymptomatic, as are the carriers of autosomal recessive disorders. Mothers of children with fragile X syndrome can have learning disorders and premature ovarian failure. Mothers of children with ornithine transcarbamylase deficiency (an X-linked disorder of nitrogen metabolism) may avoid protein in their diet and have altered mental status and vomiting episodes when they ingest a high-protein meal. Thus, although X-linked disorders may be more severe in males (including lethality in male fetuses), one should not use the gender of a patient to discredit the possibility of an X-linked disorder when the clinical presentation makes the disease plausible.
The risk of recurrence of X-linked disorders depends on the gender of the proband. Males with an X-linked condition have a 50% chance of passing the pathogenic mutations to their daughters, and no chance of passing the pathogenic mutation to their sons, because sons inherit the Y chromosome from their fathers. If a daughter inherits the pathogenic mutation, the severity of the phenotype will depend on lyonization. On the other hand, females have a 50% chance of passing the pathogenic mutation to their offspring. Once again, if the offspring is a daughter, the severity of the phenotype will depend on lyonization. All sons who inherit from their mother the X chromosome with the pathogenic mutation will manifest the disease phenotype, because males have only one X chromosome.
Not all genes on the X chromosome undergo inactivation. Some genes in the pseudoautosomal regions of the X chromosome have matching alleles on the Y chromosome. Like autosomal genes, some genes in the pseudoautosomal regions exchange genetic information during meiosis in a process called recombination or crossing-over. Other genes on the X chromosome have corresponding alleles on the Y chromosome, but do not cross over. Finally, some genes on the X chromosome are not inactivated and do not have corresponding alleles on the Y chromosome. The gene for the enzyme steroid sulfatase only exists on the X chromosome and does not undergo inactivation. Pathogenic mutations in this gene lead to a form of ichthyosis.
Mitochondrial Inheritance
Mitochondria contain approximately 16.5 kilobases of DNA (mtDNA) that encode 13 proteins that incorporate into oxidative phosphorylation complexes. (Nuclear DNA encodes most of the proteins that comprise the oxidative phosphorylation complexes.) mtDNA also encodes multiple tRNAs and two ribosomal RNAs that are essential for transcription and translation of the mtDNA. Pathogenic mutations in these genes cause many diseases, including Leigh disease; mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS); and progressive sensorineural deafness.
Mitochondria are present in every somatic human cell. They are abundant in oocytes, but the few that exist in sperm are not usually viable in the zygote. Thus, offspring inherit all of their mitochondria from their mother, and mitochondrial diseases are inherited only through the maternal lineage. A mother with a mtDNA mutation will pass the mutation to all of her offspring, but a father with a mtDNA mutation will not pass the mutation to any of his offspring. mtDNA has unique properties that make mitochondrial inheritance different from the Mendelian inheritance patterns discussed in the previous sections. Mitochondria are replicated by fission when a cell divides, and mutations can occur any time a cell divides. The mtDNA genome mutates at a rate that is ten times higher than the rate in nuclear DNA. Therefore, mtDNA mutations are much less rare than nuclear mutations, and these mutations can occur throughout the life of an individual.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


