1 Genetic Disorders and Dysmorphic Conditions
Common Chromosomal Disorders
General Principles
The Nature of Chromosomes
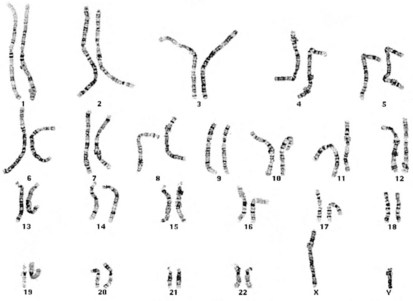
Productive insights gleaned from the results of the completed Human Genome Project have dramatically changed some of our understanding of how the human genome functions. However, it is important to introduce to the reader our current understanding of the subject matter. Human hereditary factors are located in genes (the genome). Approximately 10% are genes that encode proteins that are assembled to form tissue structures or to form enzymes that catalyze chemical reactions within cells. The other 90% have functions that are currently not clear (see also The Nature of Genes and Single-gene Disorders, later). The genes are composed of DNA and are stored in intranuclear cell organelles called chromosomes. Each chromosome contains one linear DNA molecule folded over onto itself several times, as well as ribonucleic acid (RNA) and proteins. Because all genes exist in pairs, all chromosomes must likewise exist in pairs. The members of each pair of genes are called alleles, and the members of each pair of chromosomes are known as homologues. The conventional depiction of the constitution of homologues in the nucleus is called the cell’s karyotype (Fig. 1-1). If at any gene locus the alleles are identical, that gene locus is homozygous. If the alleles are not identical, the gene locus is heterozygous.
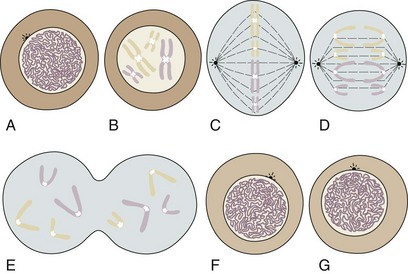
During most of a cell’s life cycle, chromosomes are diffusely spread throughout the nucleus and cannot be identified by morphologic means. Only when the cell divides does chromosome morphology become apparent (Fig. 1-2). The in vitro life cycle and the cellular division, or mitosis, of a somatic cell are illustrated in Figures 1-3 and 1-4, respectively. The life cycle and divisions, or meiosis, of a germ cell are much more complex and are not suitable for ordinary clinical evaluation.
Aneuploidy
Aneuploidy refers to an abnormality in chromosome number, in humans a chromosome number different from an even multiple of 23 (the haploid number) (Fig. 1-5). In aneuploidy there are typically 45 or 47 chromosomes instead of the usual 46. Rarely, multiples of the X or Y chromosome result in individuals with 48 or 49 chromosomes. Double aneuploidy, the simultaneous occurrence of two nondisjunctional events, has been described in the literature. In the liveborn, it usually involves one autosome and one sex chromosome. Double autosomal trisomy has been found repeatedly in spontaneous abortion but has not been demonstrated in a liveborn infant.
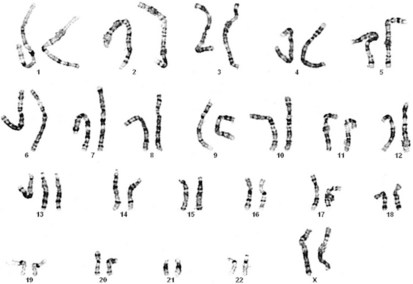
If aneuploidy occurs in a gamete as a result of an error of chromosomal division (nondisjunction or anaphase lag) during meiosis, all cells are affected in the fertilized embryo. With subsequent pregnancies, the risk for another chromosomal abnormality in the offspring is increased approximately 1% to 2% overall, in addition to the general background risk of abnormalities. The couple would be at risk for aneuploidy states of many types, not just the particular aneuploidy in their affected child. We are not yet aware of the underlying mechanism for the increased risk; however, families may benefit from an understanding of the possibilities for prenatal diagnosis in their individual case and may want to be referred for genetic counseling before the conception of another child (Fig. 1-6, A–C).
Mosaic Aneuploidy States
Mosaicism, the presence of two or more genetically different cell lines within an individual, can result from an error in division during either meiosis or mitosis. In one possible scenario aneuploidy originates during meiotic division (i.e., before conception). In such cases the fetus starts out with an aneuploid chromosomal number and, subsequently, a division error occurs, resulting in the formation of another cell line that is chromosomally normal. In other cases of mosaicism the one-celled embryo (zygote) is chromosomally normal and a division error occurs after fertilization, during mitosis of an embryonic somatic cell, resulting in aneuploidy. Most individuals with mosaicism have only two or three different lines of embryonic cells. It requires considerable laboratory investigation to distinguish the meiotic or mitotic types. Generally speaking, parents are given a 1% to 2% recurrence risk because of the possibility of mosaicism present in a parental gonad, which is not identifiable in usual tissue sample analyses (Fig. 1-7).
Hypomelanosis of Ito is characterized by marbleized or mottled areas of hypopigmented whorls of skin along the Blaschko lines and is of heterogeneous etiology. Individuals with hypomelanosis of Ito can have multiple congenital anomalies, dysmorphic features, variable mental retardation, and other neurologic findings. Karyotyping from skin lesions will reveal mosaic abnormality of chromosomes from normal or hypopigmented and hyperpigmented regions. Balanced and unbalanced chromosome aberrations and uniparental disomy may be encountered (Fig. 1-8).
Abnormalities of Chromosome Structure
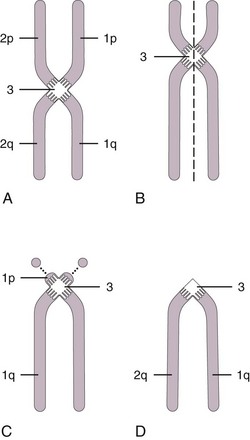
Chromosomes can be normal in number (diploid) but still be abnormal in structure. Inversions (Fig. 1-9), deletions (Fig. 1-10), and translocations (Fig. 1-11) of genetic material are examples of structural chromosomal abnormalities. These can arise as new (sporadic) mutations in the egg or sperm from which the embryo was formed, in which case the parents’ recurrence risk for another child with a chromosomal abnormality is again 1% to 2%. However, the abnormality may also be inherited from a phenotypically normal parent who is a “carrier” of a structural chromosomal abnormality (Fig. 1-12). About 1 in 520 normal individuals carries a balanced but structurally abnormal set of chromosomes, called a chromosome translocation. The term balanced, for the purposes of this chapter, means that on cytogenetic analysis the structural abnormality does not appear to have resulted in any net loss or gain of genetic material. If the apparently balanced chromosomal abnormality has been transmitted by other members of the family who are apparently phenotypically normal, it is considered a familial balanced translocation. Data suggest that a small percentage of individuals with apparently “balanced” translocations are actually mildly affected clinically by variable degrees of cognitive and physical deficits (Warburton, 1991). Thus high-resolution chromosome analyses and molecular cytogenetics techniques, such as array-CGH, are warranted in these instances including, as needed, in situ hybridization techniques using DNA probes to completely characterize the location of the chromosome breakpoints and to determine on a molecular level whether any genetic material is missing. Molecular studies for imprinting effects may also be warranted.
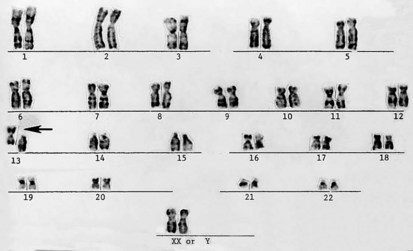
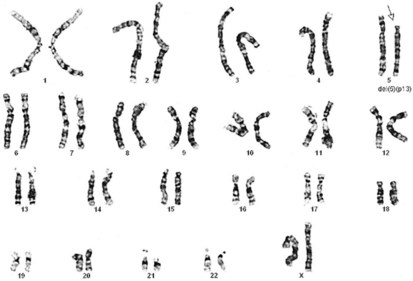
Figure 1-10 Deletion (arrow) of the p arm of chromosome 5 (cri du chat syndrome).
(Courtesy Urvashi Surti, PhD, Pittsburgh Cytogenetics Laboratory.)
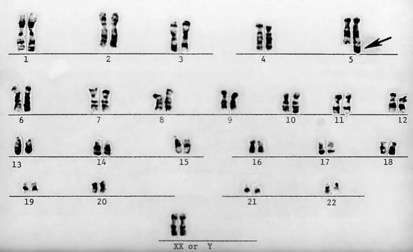
Figure 1-11 Unbalanced translocation. The additional DNA was translocated onto the q arm of chromosome 5. The abnormality was inherited from a normal carrier father (see Figure 1-12) with a balanced reciprocal translocation between the q arms of chromosome 3 and chromosome 5. The patient died of multiple birth defects and in essence had a partial trisomy of the distal portion of the q arm of chromosome 3.
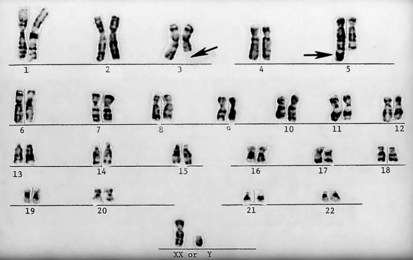
Figure 1-12 A “balanced” reciprocal translocation from chromosomes 3 to 5 in a normal man (the father of the chromosomally defective newborn whose karyotype is shown in Figure 1-11).
Incidence of Chromosomal Abnormalities
Data from Hook (1992) suggest that upward of 50% of human conceptions terminate in a spontaneous abortion. Most of these miscarriages occur so early during gestation that the pregnancy is never recognized. The earlier the abortion occurs, the more likely it is that the miscarried embryo had a chromosomal abnormality. Of recognized first-trimester abortuses, 50% are chromosomally abnormal, compared with 5% of later embryos. Among the chromosomally abnormal abortuses, the most frequent abnormalities are triploidy (69 chromosomes), trisomy 16, and 45,X (Turner syndrome) (Table 1-1). Generally speaking, triploidy and trisomy 16 are not compatible with life and are only occasionally seen among liveborn infants. Despite the fact that Turner syndrome is relatively common among liveborn infants, the majority of conceptuses with 45,X also abort spontaneously. The incidence of chromosomal abnormalities among liveborn infants in general is about 6 in 1000. Among a group including both stillborn infants and infants who die in the immediate perinatal period, the number is increased to approximately 50 in 1000.
Table 1-1 Occurrence of Chromosomal Abnormalities
| Among Spontaneous Abortuses | Incidence (%) |
|---|---|
| Overall incidence | 32.0 |
| First trimester | 52.0 |
| After first trimester | 5.8 |
| Type of abnormality seen in spontaneous abortions | |
| Trisomy 16 | |
| Other trisomies | |
| Triploidy | |
| 45,X | |
| Miscellaneous | |
| Among Liveborns | No. of Cases per 1000 |
| Overall incidence | 6.20 |
| Abnormality of autosomes | 4.19 (males and females) |
| Trisomies | |
| Balanced rearrangements | |
| Unbalanced rearrangements | |
| Abnormality of sex chromosomes | 2.03 (males and females) |
| In males: XXY, XYY, mosaics | |
| In females: 45,X (0.08), XXX, mosaics (1.43) |
About one quarter of all conceptuses are chromosomally abnormal. About 50 in 1000 stillborns have a chromosomal abnormality.
New Technologies
Fluorescence in Situ Hybridization
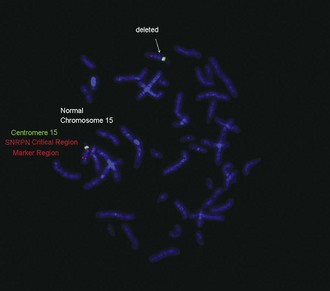
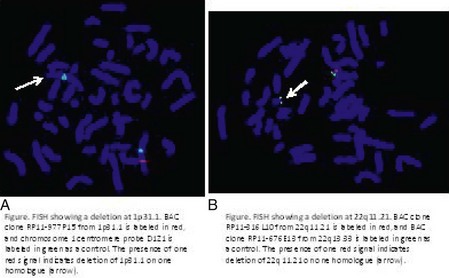
• Angelman syndrome: del 15q11-13
• Prader-Willi syndrome: del 15q11-13
• Cri du chat syndrome: del 5p15.2
• DiGeorge sequence/velocardiofacial syndrome: del 22q11.2
• Miller-Dieker/lissencephaly syndromes: del 17p13.3
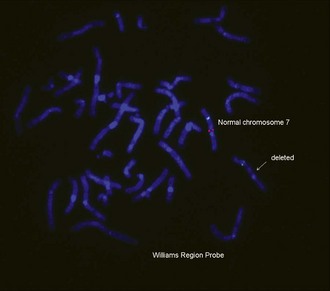
• Williams syndrome: del 7q11.23
• Smith-Magenis syndrome: del 17p11.2
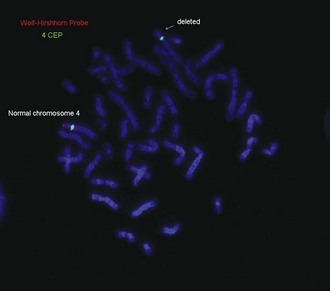
• Wolf-Hirschhorn syndrome: del 4p16.3
• Severe X-linked ichthyosis: del Xp22.3
See Figures 1-13 and 1-14 for details on Prader-Willi/Angelman syndrome and Wolf-Hirschhorn syndrome.
Array-based Technology: Microarray for Evaluation of Copy Number Variation
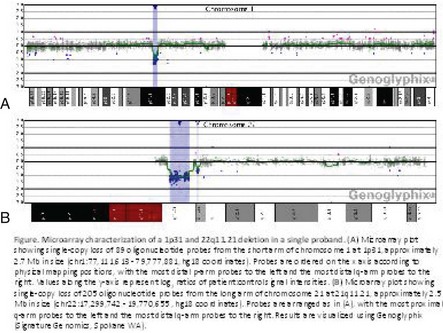
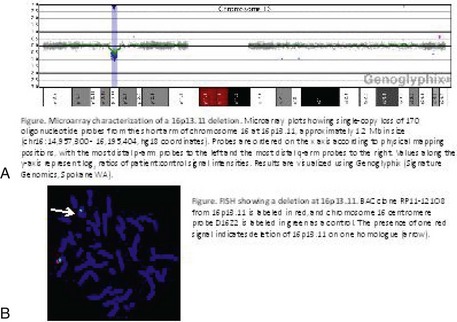
In addition to classic cytogenetics, molecular cytogenetic methods are being incorporated in clinical settings at an increased rate. More recently, conventional cytogenetics is being substituted with high-resolution molecular karyotyping using microarray-based comparative genomic hybridization (array-CGH). Array-CGH analyses are proficient in detecting imbalances in the genome and enable detection of copy-number changes at high resolution. This technique has been implemented by the American College of Medical Genetics (ACMG) as the first step in the investigation of patients with developmental delays, mental retardation, multiple congenital abnormalities, and autism spectrum disorders and has the highest diagnostic yield, up to approximately 15% to 28%. This is much higher than the diagnostic yield of G-banded karyotypes (on the order of 3%), excluding Down syndrome and other recognizable chromosomal syndrome (Miller et al, 2010). In addition, molecular cytogenetic techniques, such as array-CGH, have demonstrated that approximately 20% of apparently balanced chromosome translocations, de novo or familial, have gain or loss of genetic material at the breakpoints. Therefore, molecular cytogenetic studies are warranted because they completely characterize the location of the chromosome breakpoints and potentially identify additional genetic material that may be duplicated or deleted that would not otherwise be detected by the traditional cytogenetic methods. FISH and other molecular techniques are now used primarily to confirm the imbalances detected by array-CGH. With microarray testing, many new microdeletion and microduplication syndromes have emerged (e.g., deletion 1p36, deletion 1q21.1, and deletion 16p13.11 syndromes) (Fig. 1-15; and see e-Figs. 1-1 through 1-3).
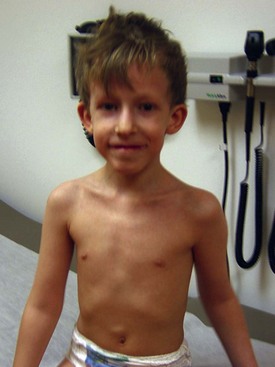
DiGeorge sequence is discussed in Chapter 4, Williams syndrome is discussed in Chapter 5, and Angelman and Prader-Willi syndromes are covered later in this chapter. The remaining syndromes are outlined briefly in Table 1-2 and in Figure 1-16, A–D; Figure 1-17; and Figure 1-18.
Table 1-2 Some Syndromes Identifiable with Fluorescence in Situ Hybridization Probes
| Syndrome | Major Findings | Comments |
|---|---|---|
| Cri du chat (deletion 5p15.2) | Microcephaly, round face, down-slanting palpebral fissures, epicanthal folds, hypertelorism, catlike cry in infancy | |
| Isolated lissencephaly | Lissencephaly (incomplete development of brain with smooth surface) | Approximately 30% have deletion 17p13.3 |
| Miller-Dieker phenotype with lissencephaly | Microcephaly, lissencephaly, variable high forehead, vertical furrowing of central forehead, low-set ears, small nose with anteverted nostrils, congenital heart disease, poor feeding | Deletion 17p13.3 in vast majority |
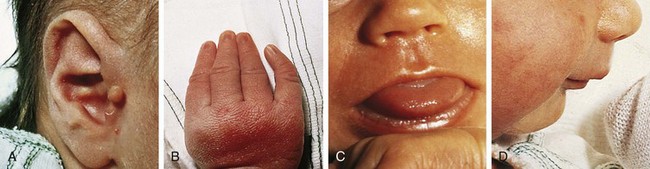
| Deletion 22q11.2 | Phenotypes: | Appears to be a common deletion and should be considered in the differential diagnosis of children with multiple anomalies even if the features are not classic to any one phenotype |
| Wolf-Hirschhorn (deletion 4p16.3) | Moderate to severe cognitive impairment, hypertelorism, preauricular pit or tag, broad nasal bridge, micrognathia, cleft palate, short philtrum, growth deficiency | |
| Smith-Magenis (deletion 17p11.2) | Brachycephaly, flat facies, broad nasal bridge, short stature | Self-hugging behaviors, sleep disturbances |
Approach to the Evaluation of a Dysmorphic Child
Approximately 2% to 3% of liveborn infants have an observable structural abnormality. This number rises to about 4% to 5% by the time the child is old enough to attend school. Structural differences can be determined to be either major or minor in character (Table 1-3, and Figs. 1-19 and 1-20). Major structural anomalies have functional significance. Examples are polydactyly, colobomas of the iris (see Chapter 19, Fig. 19-69), meningomyelocele, and cleft lip. Minor anomalies are usually of cosmetic importance only. Examples are epicanthal folds of the eyes, single transverse palmar creases, and supernumerary nipples. The incidence of isolated major anomalies in the general newborn population is approximately 1%, and the incidence of minor anomalies is approximately 14%. Both are more common in premature newborns.
Table 1-3 Examples of Congenital Anomalies
| Category | Major | Minor |
|---|---|---|
| Craniofacial | Choanal atresia | Plagiocephaly |
| Flat occiput | ||
| Eyes | Coloboma of iris | Epicanthal folds |
| Ears | Microtia | Preauricular pit |
| Hands | Polydactyly Absent thumbs | Single transverse palmar crease |
| Clinodactyly |
Definitions of the classifications of structural anomalies aid in communication between clinicians and in the process of evaluation and are summarized from Jones (2006):
Malformation: A malformation is an abnormality of embryonic morphogenesis of tissue. It usually results from genetic, chromosomal, or teratogenic influences, but it can be of multifactorial etiology. Malformations are divided into two main categories: those that constitute a single primary defect in development and those that represent a single component of a multiple malformation syndrome. A multiple malformation syndrome can be defined as one having several observed structural defects in development involving multiple organ systems that share the same known or presumed etiology. Malformations often require surgical intervention.
Deformation: A deformation represents an alteration (often molding) of an intrinsically normal tissue caused by exposure to unusual extrinsic forces. A classic example is clubfoot, which may be the result of uterine constraint from crowding associated with a multiple gestation. A more severe example is the compressed facial features (“Potter facies”) of a child exposed to severe uterine constraint associated with oligohydramnios, due to renal agenesis (see Chapter 13, Fig. 13-38). The vast majority of deformations respond to medical therapy alone and have a relatively good prognosis in contrast to malformations, which frequently require surgical intervention.
Disruption: A disruption represents a breakdown of normally formed tissue; the breakdown may be the result of vascular accidents or exposure to adverse mechanical forces that are usually more severe than those that produce deformation. A classic example is the combination of clefting, constriction bands, and limb reduction defects associated with the presence of amniotic bands (see Chapter 2, Fig. 2-46). The earlier these vascular accidents or abnormal forces occur during embryogenesis, the more severe the resulting defects (Fig. 1-21).
Dysplasia: Dysplasia is characterized by abnormal organization of cells within tissue, which usually has a genetic basis. An example is achondroplasia, the most frequent form of skeletal dysplasia.
(Note: Each of the preceding categories can have a sequence [see below] associated with it.)
Sequence: The term sequence refers to a recognizable pattern of multiple anomalies that occurs when a single problem in morphogenesis cascades, resulting in secondary and tertiary errors in morphogenesis and a corresponding series of structural alterations. A classic example is the Robin malformation or Pierre Robin sequence, in which the single primary malformation is microretrognathia (see Chapter 23, Fig. 23-63). The resulting glossoptosis, or posterior placement of the tongue in the oropharynx, interferes with normal palatal closure if the lingual displacement occurs before 9 weeks’ gestation. The resulting cleft palate is U-shaped, rather than having the V shape that is usually seen in classic cleft palate, a finding that aids in recognition.
Association: An association is a pattern of malformations that occurs together too frequently to be due to random chance alone, but for which no specific etiology is yet recognized.
The history should include the following:
• Course of the pregnancy, complications including possible infections or environmental exposures, medications/substance abuse
• Prior pregnancies, spontaneous abortions, stillborns, or infant/child deaths for this couple
• Labor/delivery/perinatal problems
• Meticulous family history with family tree going back three generations and including the following:
The physical examination entails the following:
Abnormalities of Autosomes
Down Syndrome
No single physical stigma of Down syndrome exists; rather, the clinical diagnosis rests on finding a recognizable constellation of clinical characteristics including a combination of major and minor anomalies (Fig. 1-22).
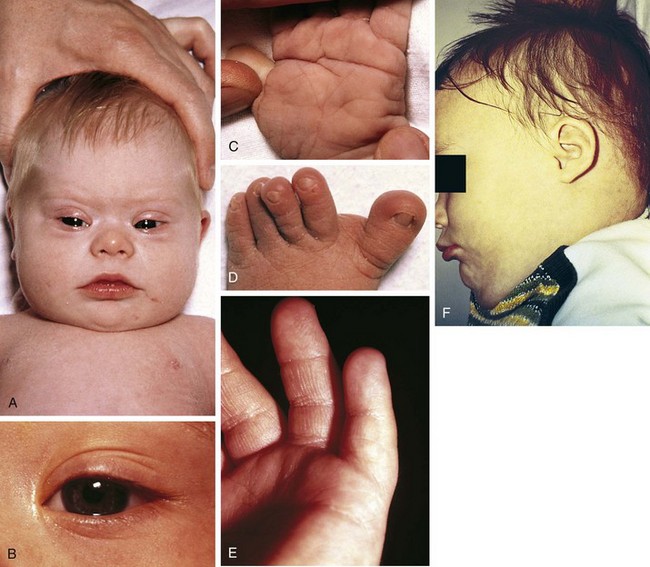
The etiology of Down syndrome is trisomy 21, the presence of an extra chromosome 21 either as a simple trisomy or as part of a chromosome 21 fused with another chromosome. These fused chromosomes are often robertsonian translocation chromosomes or isochromosomes. Cases of mosaicism, in which trisomy 21 cell lines coexist with cell lines with the standard 46 chromosomes, exist as well and may range in phenotype from normal to that typical of complete trisomy 21. An association between trisomy 21 and advanced maternal age is clear (Table 1-4).
Table 1-4 Maternal Age–Specific Risk for Trisomy 21 at Live Birth
| Maternal Age (yr) | Prevalence at Live Birth |
|---|---|
| 25 | 1/1350 |
| 30 | 1/890 |
| 35 | 1/355 |
| 40 | 1/97 |
| 45 | 1/23 |
Trisomy 13
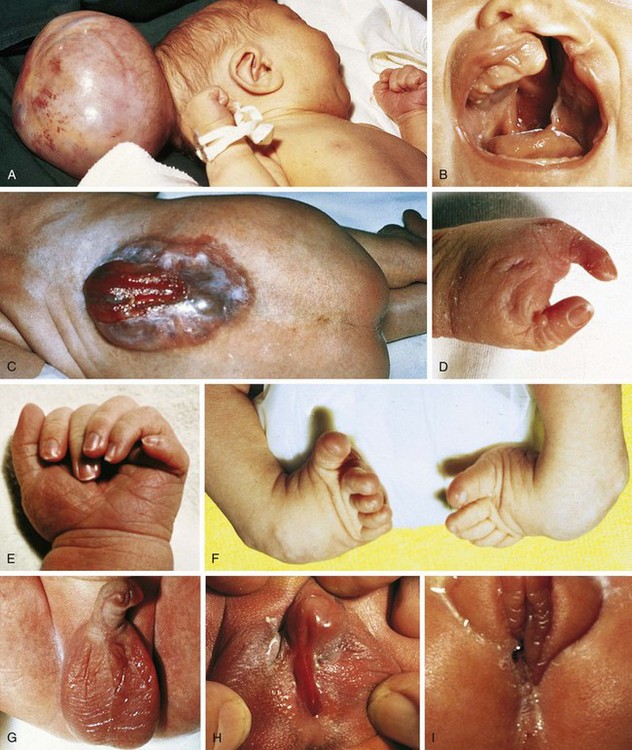
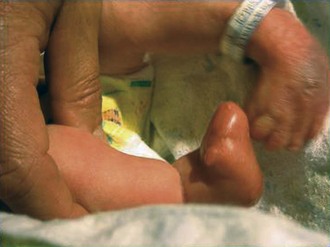
Trisomy 13 is a relatively rare (1 in 5000) genetic condition caused by the presence of additional chromosome material from all or a large part of chromosome 13. The vast majority of embryos with classic trisomy for a complete 13th chromosome abort spontaneously, but approximately 5% survive to be liveborn. They have a severe, recognizable pattern of malformation that allows clinicians to suspect this etiology immediately (Fig. 1-23). The hallmark features are defects of forebrain development related to those seen in holoprosencephaly, aplasia cutis congenita, polydactyly (most frequently of the postaxial type), and narrow hyperconvex nails. A broader listing of features is outlined in Table 1-5, which can be useful in comparing the features frequently seen in infants with trisomy 13 with those seen in trisomy 18. As with many syndromes, trisomy 13 and trisomy 18 share structural abnormalities; however, they usually are distinguishable on the basis of the pattern of anomalies present. Liveborn infants with trisomy 13 represent those who have the least severe structural abnormalities of major organs. Of these, about 5% survive the first 6 months of life. Thus discussions with parents about surgical interventions must take into account the small possibility of long-term survival and require sensitivity to the needs of the child and family.
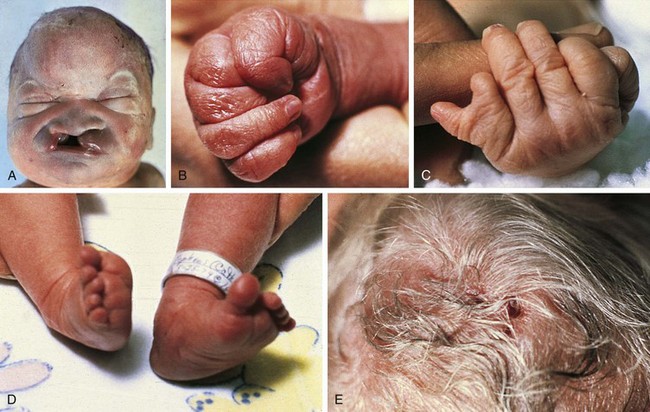
Table 1-5 Physical Abnormalities and Frequencies of Occurrence in Trisomy 13 and Trisomy 18 Syndromes
| Abnormality | Trisomy 13 | Trisomy 18 |
|---|---|---|
| Severe developmental retardation | †††† | †††† |
| Approximately 90% die within first year | †††† | †††† |
| Cryptorchidism in males | †††† | †††† |
| Low-set, malformed ears | †††† | †††† |
| Multiple major congenital anomalies | †††† | †††† |
| Prominent occiput | † | †††† |
| Cleft lip and/or palate | ††† | † |
| Micrognathia | †† | ††† |
| Microphthalmos | ††† | †† |
| Coloboma of iris | ††† | † |
| Short sternum | † | ††† |
| Rocker-bottom feet | †† | ††† |
| Congenital heart disease | †† | †††† |
| Scalp defects | ††† | † |
| Flexion deformities of fingers | †† | †††† |
| Polydactyly | ††† | † |
| Hypoplasia of nails | †† | ††† |
| Hypertonia in infancy | † | ††† |
| Apneic spells in infancy | ††† | † |
| Midline brain defects | ††† | † |
| Horseshoe kidneys | † | ††† |
Symbols: Relative frequency of occurrence ranges from †††† (usual) to † (rare).
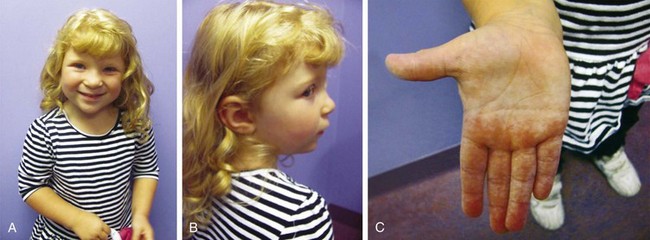
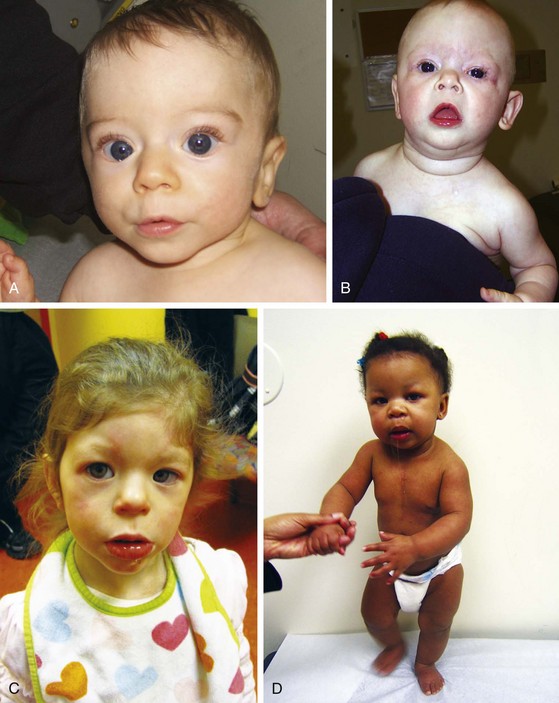
Trisomy 18
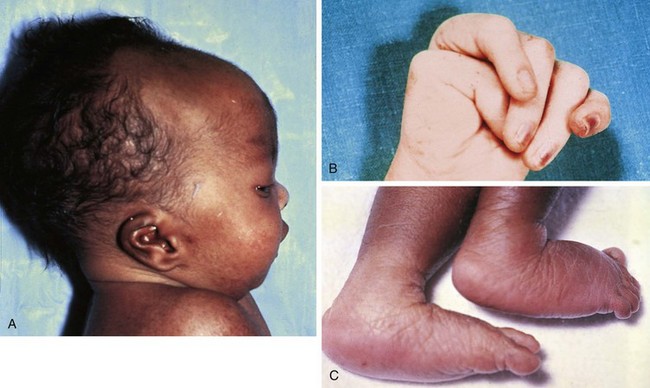
The chromosomal disorder trisomy 18 occurs in approximately 3 in 10,000 newborns, and females are more likely to be liveborn. Affected infants are small for gestational age and have a frail appearance, and the face tends to appear petite relative to the rest of the craniofacial contour (Fig. 1-24, A). They also have a recognizable pattern of malformation, but in these infants hallmark features—clenched hands with overlapping fingers (see Fig. 1-24, B), short sternum, and “low arch” fingerprint patterns—are minor anomalies. Major anomalies, especially congenital heart disease, are generally present as well and are the source of significant morbidity and mortality. Other common findings include a prominent occiput, low-set and structurally abnormal ears, micrognathia, and rocker-bottom feet (see Fig. 1-24, C). See Table 1-5 for a broader listing of clinical features that can be useful in distinguishing trisomy 18 from trisomy 13, which shares many of the same structural abnormalities.
Abnormalities of Sex Chromosomes
Turner Syndrome
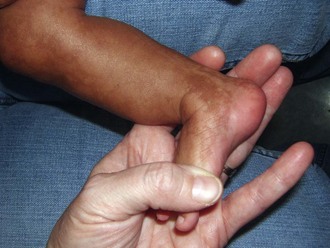
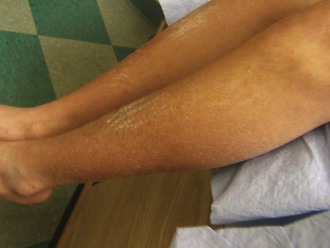
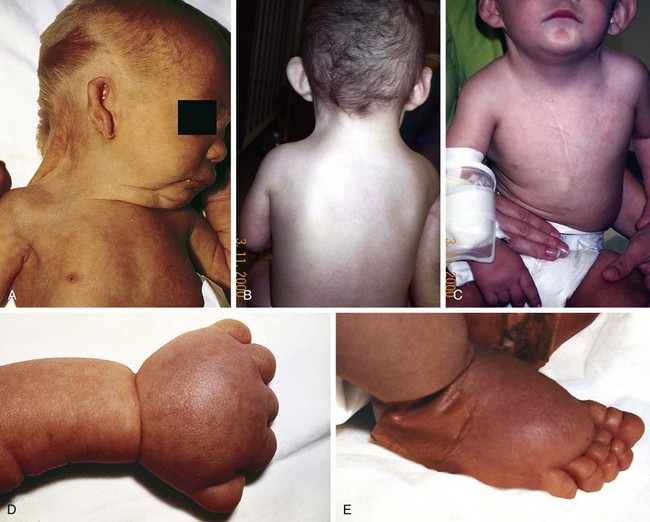
Turner syndrome is one of the three most common chromosomal abnormalities found in early spontaneous abortions. The phenotype is female. About 1 in 2000 liveborn females has Turner syndrome. Primary amenorrhea, sterility, sparse pubic and axillary hair, underdeveloped breasts, and short stature (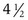 to 5 ft) are the usual manifestations. Other external physical features may include webbing of the neck; cubitus valgus; a low-set posterior hairline; a shield chest with widely spaced nipples; and malformed, often protruding, ears (Fig. 1-25, A–E). Internally, renal anomalies may be present along with congenital heart disease, particularly bicuspid aortic valve (in 30% of cases) and coarctation of the aorta (in 10% of cases). Affected women have an infantile uterus, and their ovaries consist only of strands of fibrous connective tissue. Newborns often have lymphedema of the feet and/or hands (Fig. 1-25, D and E), which can reappear briefly during adolescence. Mental development is usually normal. Schooling and behavioral problems seem to be the same as in age-matched control subjects, although difficulty with spatial orientation such as map reading may be a problem. The classic physical findings of Turner syndrome may be absent, or the abnormalities may be so minimal in the newborn that the diagnosis is missed. The first indication may be unexplained short stature in later childhood or failure to develop secondary sex characteristics by late adolescence. Thus a chromosome study is indicated as part of the diagnostic workup of adolescent girls with these complaints.
to 5 ft) are the usual manifestations. Other external physical features may include webbing of the neck; cubitus valgus; a low-set posterior hairline; a shield chest with widely spaced nipples; and malformed, often protruding, ears (Fig. 1-25, A–E). Internally, renal anomalies may be present along with congenital heart disease, particularly bicuspid aortic valve (in 30% of cases) and coarctation of the aorta (in 10% of cases). Affected women have an infantile uterus, and their ovaries consist only of strands of fibrous connective tissue. Newborns often have lymphedema of the feet and/or hands (Fig. 1-25, D and E), which can reappear briefly during adolescence. Mental development is usually normal. Schooling and behavioral problems seem to be the same as in age-matched control subjects, although difficulty with spatial orientation such as map reading may be a problem. The classic physical findings of Turner syndrome may be absent, or the abnormalities may be so minimal in the newborn that the diagnosis is missed. The first indication may be unexplained short stature in later childhood or failure to develop secondary sex characteristics by late adolescence. Thus a chromosome study is indicated as part of the diagnostic workup of adolescent girls with these complaints.
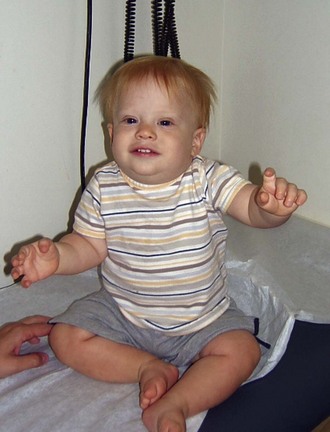
Klinefelter Syndrome
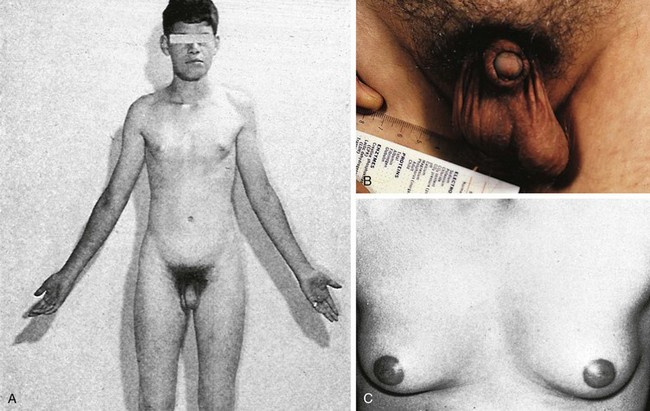
One in 500 newborn boys has Klinefelter syndrome. The physical stigmata are subtle and usually not obvious until puberty, at which time the normal onset of spermatogenesis is blocked by the presence of two X chromosomes. Consequently the germ cells die, the seminiferous tubules become hyalinized and scarred, and the testes become small. Testosterone levels are below normal adult male levels, although the level varies from case to case (the average being about half as much as normal). Hence there is a wide range in degree of virilization. At one extreme is the man with a small penis and gynecomastia (Fig. 1-26); at the opposite extreme is the virile mesomorph with a normal penis. Scoliosis may develop during adolescence. The average full-scale IQ of men with Klinefelter syndrome is 98, which is about the same as the general population. Behavioral problems may be more common than in the population at large, however.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


