Schematic diagram showing the timeline for embryologic and fetal lung development, with pictorial representation below.
At birth
The amniotic fluid and lung secretions fill the airways prior to birth. During early labor, the production of fetal adrenaline and maternal thyrotrophin-releasing hormone cause the fetal pulmonary epithelial cells to absorb lung fluid. Active pulmonary fluid absorption is increased and most of the lung fluid is cleared within the first 2 hrs of extrauterine life. There is active transport of sodium ions from the alveolar spaces into the interstitial tissues. Tactile stimulation and changes in temperature are potent stimulants to fetal breathing. The inflation of the lungs leads to a fall in pulmonary vascular arteriolar resistance and increased pulmonary blood flow. The alveoli become aerated and are lined with surfactant, and the lungs become much easier to aereate at lower negative intrathoracic pressures.
Normal healthy lungs are composed of an orderly system of tubes (airways) and sacs (airspaces or alveoli) in a strict relationship to pulmonary blood vessels (arterial from the right ventricle and venous return to the left atrium) with a normally developed systemic blood supply (aorta to superior vena cava) and lymphatic drainage. Congenital lung malformations arise whenever one or more of these structures are abnormal or when their relationships are altered.
Congenital lung malformations represent 5–19% of all congenital anomalies. This range may be an underestimate because of the high frequency of undetected or asymptomatic lesions.
Ultrasound of the normal thorax
The main views used to assess the thorax are the four-chamber view of the heart and the saggital view of the diaphragm. The heart usually occupies about one-third of the chest and the axis should be about 45° to the anteroposterior midline. The position within the chest should also be noted to identify any mediastinal shift. Other important observations include the presence of polyhydramnios, which may reflect a mass or pressure effect if a lung lesion, mass or abnormality is identified.
Ultrasound of thoracic malformations
Lung malformations are rare but are an important cause of morbidity and mortality from infection, hemorrhage and hypoxia. The overall outcome is dependent on their size, local pressure effects and effect on lung function[1]. Small lesions are often asymptomatic and easily missed. Most fetal parenchymal lung pathology appears as an echogenic lung mass, which may be unilateral (congenital diaphragmatic hernia (CDH), congenital cystic adenomatoid malformation of the lung (CCAML), pulmonary sequestration) or bilateral (laryngeal atresia), but lesions may apparently resolve later in the pregnancy or be complicated by cystic components often seen in CDH and CCAML. Large fluid-filled lobes may be present in congenital lobar emphysema. Indeed postnatal X-rays and ultrasound scans may appear normal and computed tomography (CT) or magnetic resonance imaging (MRI) may be indicated.
In the case of pulmonary hypoplasia, there may be other clues to their aetiology, such as oligohydramnios or renal pathology as the principle etiology, ultrasound markers for fetal chromosomal abnormalities, skeletal dysplasia or central nervous system malformations, or abnormal movements or posture due to neuromuscular disease.
Lung biometry and blood flow studies
Lung growth is linear from about 16–40 weeks.
Ratios between thoracic circumference (TC) or diameter and reference measurements such as abdominal circumference (AC), head circumference, femur length and end-diastolic biventricular cardiac diameter remain constant with a high correlation coefficient in normal pregnancy[2–4].
The TC:AC ratio is normally >0.8 after 20 weeks; absolute and relative TC measurements are useful in predicting subsequent pulmonary hypoplasia and serial TC:AC ratios are predictive in early premature rupture of membranes[5].
Lung:head ratios (LHR) have also been shown to correlate with subsequent prognosis[6].
LHR ≤1, the prognosis is poor.
LHR 1.0–1.4, extracorporeal membranous oxygenation (ECMO) is often needed.
LHR >1.4, the prognosis is better.
Prenatal MRI is a useful adjunct to ultrasound imaging and also allows assessment of the nature and the size of lungs and lung lesions. Whilst pulmonary artery blood flow can be measured using Doppler ultrasound, it is of limited value in assessing the likelihood pulmonary hypoplasia, although color Doppler imaging is useful in differentiating CCAML from pulmonary sequestration.
Congenital diaphragmatic hernia
Congenital diaphragmatic hernia (CDH) affects around 1:4,000 births and occurs when the diaphragm fails to form a complete barrier in the pleuroperitoneal canal between the thoracic and abdominal cavities (usually around 10 weeks). This leads to the presence of any combination of bowel, stomach and other herniated organs into the thoracic cavity causing significant morbidity and mortality from the compounded effects of pulmonary hypoplasia, abnormal lung development, pulmonary hypertension and cardiac compression.
There is a high incidence of associated abnormalities (30–60%), chromosomal abnormalities (10–20%) and other genetic syndromes (e.g., Beckwith–Wiedemann syndrome), and thus a careful search for other anomalies and the offer of karyotyping is appropriate. CDH is often identified at the routine 20-week anomaly scan when the fetal stomach cannot be found in its normal position in the abdomen below the diaphragm. The stomach and other herniated viscera, such as the bowel, liver and spleen, are found in the chest. Some CDHs are variable in appearance with the stomach and bowel sliding in and out of the chest. Other differential diagnoses include CCAML, bronchopulmonary sequestration and thoracic teratomas.
Pressure effects within the chest can cause esophageal compression leading to polyhydramnios and cardiac compression leading to hydrops. Thus, in all cases of CDH, serial scans are required to monitor for these features.
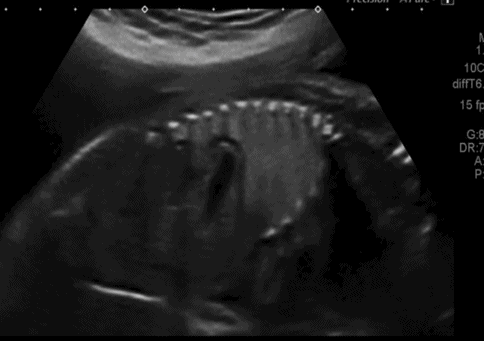
Saggital ultrasound scan section through fetal trunk showing congenital diaphragmatic hernia, with the stomach clearly seen within the thoracic cavity.
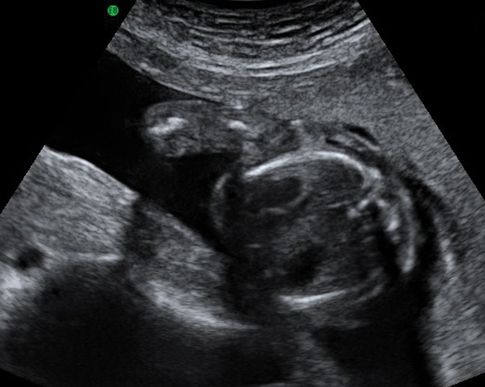
Transverse ultrasound scan through fetal thorax showing fetal heart and stomach in the same plane. Note the appearance of a single rib on either side confirms that this is not an oblique view.
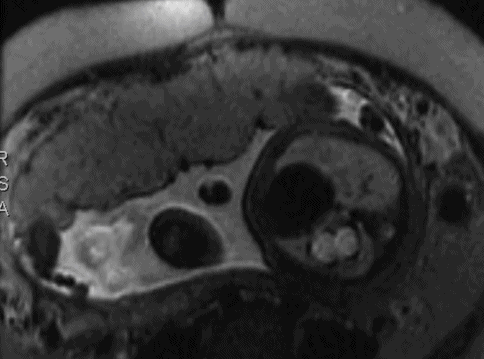
Transverse magnetic resonance imaging of left- sided congenital diaphragmatic hernia with the stomach seen in the middle of the left hemithorax. The right lung is normal. Axial, sagittal and coronal T2-weighted sequences were obtained through the fetal thorax and abdomen. The stomach and several loops of bowel are lying posteriorly within the lower left thorax. On the sagittal images, there is suggestion that the anterior part of the diaphragm is intact but cannot differentiate between this being a posterior diaphragmatic hernia or an eventration. There is, however, a good lung volume bilaterally and the heart lies centrally with no mediastinal shift. Normal abdominal organs.
Prognosis
Various factors have been assessed to try to predict the prognosis in cases of CDH, with liver herniation a predictor of poor outcome and LHR ratio being shown to be of prognostic value[6]. The LHR is measured at the level of the heart in the four-chamber view and compared with the HC. Low LHR values of <1 are associated with poor survival rates around 10%. The LHR is gestation dependent and gestation specific charts should be used rather than a fixed threshold value.
Attempts to assess lung volume using two-(2D) and three-dimensional (3D) ultrasound and MRI have had variable success.
Initial management
Following a diagnosis of CDH, referral should be made to a tertiary fetal medicine unit for a detailed anatomic survey, including an assessment of fetal anatomy, identification of ultrasound markers, amniotic fluid index, a fetal echo, karyotyping and more recently microarray-comparative genome hybridization studies. Amniotic fluid is preferred to fetal blood as conditions like Pallister–Killian syndrome can be missed by analyzing fetal blood. Fetal MRI or 3D ultrasound may be of value to evaluate lung volume. Counseling should be offered with involvement of a multidisciplinary team, including a pediatric surgeon and/or a neonatologist regarding the neonatal and long-term implications, and neonatal and surgical management. Termination of pregnancy would also be an option and should be discussed in view of the high mortality and morbidity. Subsequent management includes serial scans to identify polyhydramnios, which increases the risk of preterm labor and may require specific management, and for fetal hydrops, which may warrant early delivery.
In utero treatment
Treatment in utero has been successful in some cases using ultrasound and then fetoscopic-guided tracheal occlusion. This technique has been shown to improve lung mass, airway branching and pulmonary vascular development. The main benefit seems to be preventing the escape of lung fluid, which distends the airways, and importantly the distal airways, during the critical canalicular and secular phases of lung development. For this reason, it is of minimal value in late pregnancy. Although the evidence from randomized controlled trials has shown little benefit in terms of overall survival from in utero treatment (73% treated in utero compared to 77% controls), a subgroup with potentially the worst prognosis who had liver herniation and LHR <1 did show a marked improvement in survival from 10% to 40–50% when treated in utero[7]. An unresolved challenge is the morbidity and mortality of premature delivery.
Delivery and neonatal care
In general, delivery would be at term in a tertiary unit with access to on-site neonatal surgery and neonatal intensive care unit (NICU) facilities. A senior pediatrician should be present with early elective intubation. There is no advantage to cesarean section and the mode of delivery would be on the basis of normal obstetric grounds. Following delivery, stabilisation is essential prior to surgery with major challenges from pulmonary hypoplasia and pulmonary hypertension. In some cases, ECMO may need to be considered. Prostacyclin and nitrous oxide may also be of benefit. Surgery will usually be with primary closure or in cases of a large defect, a mesh may be required. Resumption of normal feeding is often slow, and prolonged total parenteral nutrition may often be required.
Congenital cystic adenomatoid malformation of the lung
Cystic congenital adenomatoid malformation of the lung (CCAML) affects 1 in 1,100–3,500 live births, is more common in males than females and accounts for about 25% of all lung malformations. Abnormal lung development is thought to be due to aberrant HOXB-5, FGF-7 and PDGFB genes. The affected parts of the lung are deficient in normal alveoli, and there is both proliferation and abnormal branching and cystic dilatation of the terminal respiratory bronchioles. The cysts that result from the overgrowth of the terminal bronchioles may be small and give an echogenic appearance (microcystic) or may coalesce into larger visible cysts (macrocystic), which can be seen on ultrasound.
Most are unilateral (85%) and they are commoner on the left side (60%). CCAML will often be identified at the routine 20-week anomaly scan as a unilateral or bilateral echogenic lung mass with or without visible cysts. Local pressure effects can occur causing pulmonary hypoplasia of the surrounding and indeed contralateral lung and the risk of esophageal compression and polyhydramnios and cardiac compression and hydrops. The risk of hydrops is greatest for those with large lesions and high CCAML:HC ratios have been shown to be predictive of subsequent development of hydrops. Generally, the maximum growth is in midtrimester, with reduced growth towards the end of the pregnancy. The echogenicity is also often reduced in late pregnancy and the lesion may no longer be visible on ultrasound giving the false reassurance that it has resolved.
Differentiation from bronchopulmonary sequestration may be difficult as the vascular supply and drainage is derived from the pulmonary system and lesions may have a systemic arterial blood supply similar to that seen in bronchopulmonary sequestration. These are referred to as hybrid lesions. Some authorities suggest that since it is not possible to distinguish CCAML from these hybrid lesions, the term “congenital pulmonary airway malformation” (CPAM) should be used instead.
After birth, the ventilation of the abnormal alveolar tissue, which can often be seen on a chest X-ray, poses a significant risk of respiratory distress and pneumothorax. Unilateral solid or cystic lung masses on a neonatal X-ray should always arouse suspicion of a possible CCAML.
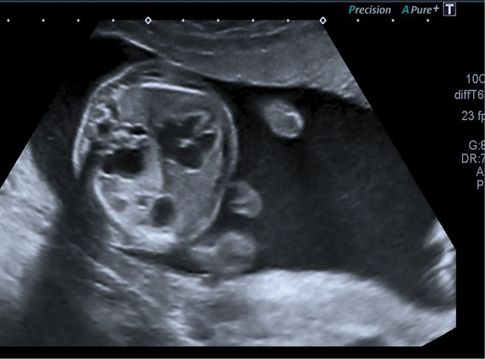
Transverse ultrasound scan section through fetal thorax showing extensive macrocystic congenital cystic adenomatoid malformation of the lung, echogenic lung and large lung cysts on the right side.
Ultarsound saggital section through fetal body showing extensive macrocystic congenital cystic adenomatoid malformation of the lung and echogenic lung.
Prognosis
In general, about 45–85% of antenatally diagnosed CCAML will spontaneously regress; however, large macrocystic or solid lesions can cause local pressure effects leading to polyhydramnios and preterm delivery, hydrops, pulmonary hypoplasia, cardiac dysfunction and perinatal death. Assessment of CPAM volume ratio (CVR), based on comparison with HC on ultrasound and fetal MRI, has been shown to be prognostic with smaller lesions with a CVR <1.6 having a better outcome[8–10].
Classically there are three described types of CCAML.
(1) Macrocystic (13%). Best prognosis. One or more large cysts (>5 mm) lined with pseudostratified ciliated epithelium.
(2) Microcystic (73%). Small cysts giving the lungs a bright echogenic appearance, lined with ciliated columnar or cuboidal epithelium.
(3) Solid (13%). Worst prognosis. Cuboidal epithelium-lined bronchioles. This tissue is airless in postnatal life.
Although the macrocystic types have larger cysts, they tend to involve smaller volumes of lung than the microcystic or solid types, and hence their relatively better prognosis. Other pathologic classifications are based on a five-category system:
(1) type 0 – acinar dysplasia
(2) type I – multiple large cysts or a single dominant cyst
(3) type II – multiple evenly spaced small cysts – may be associated with other abnormalities
(4) type III – mainly solid mass
(5) type IV – mainly peripheral cysts.
It is thought that the appearance may relate to the abnormal phase of lung development with types I–III having bronchiolar epithelium due to abnormal pseudoglandular development and type IV having alveolar acinar epithelium. All types are usually in direct communication with the tracheobronchiolar tree.
Antenatal diagnosis and initial management
The main features of CCAML are often identified on the routine anomaly scan on the basis of either an echogenic lung mass or cystic areas within the lungs. More subtle findings, such as cardiac axis deviation or mediastinal shift, should prompt a careful search for other lung pathology, such as CCAML or CDH.
CCAML is usually an isolated abnormality but some may be associated with other structural abnormalities, particularly cardiac (tetralogy of Fallot (TOF) and truncus arteriosus). A detailed anatomic survey should therefore be undertaken including an assessment of fetal anatomy, amniotic fluid index and exclusion of hydrops and a fetal echo. The role for karyotyping is controversial as this is not usually a feature of CCAML, but the suspected diagnosis of CCAML may be incorrect. Following the diagnosis of any significant lung pathology, the patient should be referred to a tertiary fetal medicine center for multidisciplinary review, and subsequent management may include counseling by a pediatric surgeon and/or a neonatologist regarding the neonatal and long-term implications, and neonatal and surgical management. Termination of pregnancy would also be an option, particularly for larger or complex nonisolated lesions, and should also be discussed. A careful search for a feeding vessel, which would be suggestive of a bronchopulmonary sequestration as opposed to a hybrid lesion should be sought, but in hybrid cases they tend to arise from the descending aorta. Other differential diagnoses include a CDH, bronchogenic cyst or neuroblastoma.
Subsequent management
Subsequent management includes serial scans to identify polyhydramnios, which increases the risk of preterm labor and may require specific management, and fetal hydrops, which may warrant early delivery. The echogenicity of the lesions usually darkens in late pregnancy giving the false reassurance that the CCAML has apparently resolved. Mediastinal shift may be an indicator of a higher risk of pulmonary hypoplasia and respiratory failure.
In utero treatment
In type 1 or macrocystic CCAML, there may a single large cyst, which can cause significant pressure effects leading to polyhydramnios or hydrops. Pleuroperitoneal aspiration or drainage may be beneficial, particularly when hydrops is present. In such cases, shunting may be preferable as the fluid tends to reaccumulate following aspiration.
Delivery and neonatal care
In most cases, the multidisciplinary team is likely to advise delivery at term in a tertiary unit with access to on-site neonatal surgery and NICU facilities as antenatal assessment cannot accurately predict the likely degree of respiratory compromise in the newborn, although larger lesions and those with significant mediastinal shift pose a greater risk. A senior pediatrician should be present with recourse to early elective intubation. There is no advantage to cesarean section, and the mode of delivery would be on the basis of normal obstetric grounds. Admission to NICU for observation is advised and an early chest X-ray should be performed. In most cases (about 60%), cystic adenomatoid malformation manifests clinically with respiratory complications soon after the neonatal period. It also results in recurrent chest infections due to poor clearance of mucous and other lung secretions.
Many lesions will regress to some extent following birth but late effects, though rare, include malignancy in the form of pulmonary blastoma, rhabdomyosarcoma, and bronchoalveolar carcinoma. Whilst surgery may usually be deferred until about 2 years old, if the baby is well and there is no significant respiratory compromise or recurrent serious respiratory infections, because of the risk of recurrent infections and malignancy, the affected lung tissue is usually removed and the long-term outcome is then usually good. Early surgery may, however, sometimes be needed if there is significant early respiratory compromise.
Bronchopulmonary sequestration
Pulmonary sequestration accounts for 6% of all congenital lung malformations and affects 1:1,000 live births. It mostly affects the lower lobes. A sequestration is a bronchopulmonary mass without a normal bronchial communication, and is thought to be due to development of a primitive lung bud from the foregut during embryonic development. The affected part of the lung is not connected to the airways, although rarely, communication with the tracheobronchial tree may occur via fistulas. The vascular supply is usually also abnormal with the arterial blood supply coming from the systemic side of the circulation (and directly off the aorta in 80% of cases) rather than the pulmonary circulation. It is important to distinguish sequestration from Scimitar syndrome in which there is tracheobronchial communication and venous drainage.
The affected part of the lung may be intralobar (75%) or extralobar (25%) with no sex differentiation in the incidence of intralobar lesions but a 4:1 male-to-female ratio in those with extralobar lesions[11]. It is thought that intralobar lesions are due to abnormal early lung budding, which leads to an area of lung with no separate pleural covering, whereas extralobar sequestration forms a lung lesion consisting of lung parenchymal tissue with a distinct pleural covering, which delineates it from the normal surrounding lung.
Intralobar lesions
Intralobar or intrapulmonary lesions more commonly affect the posterior lung bases (99%). About 10% are associated with other structural abnormalities, such as CDH and eventration, tracheoesophageal fistula, cardiac defects, foregut abnormalities and chromosomal abnormalities. Despite the arterial supply being from the systemic circulation, the venous drainage is usually through the pulmonary circulation. One important differential diagnosis is an azygous lobe, which is a malformation of the right upper lobe caused by an aberrant azygous vein suspended by a pleural mesentery. An azygous lobe is a radiographic curiosity without clinical significance that occurs in 0.5% of the general population.
Extralobar lesions
Although most (90%) extralobar (extrapulmonary) lesions are above the diaphragm and on the left side (80%), they can also occur below the diaphragm and thus can be easily missed or misinterpreted. Associated structural abnormalities are more common than for intralobar lesions affecting over 60% of cases and may include CDH, gut duplication, vertebral abnormalities and pulmonary hypoplasia. Venous drainage is usually into the systemic circulation via the subdiaphragmatic veins via the azygous system into the right atrium. Pleural effusions may also be present in up to 10%, thought to be due to increased lymphatic transudate in the thorax. They may also connect with the gut.
Imaging
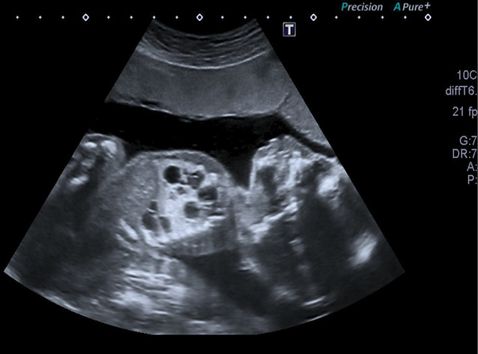
Transverse ultrasound scan section through fetal thorax showing intralobar bronchopulmonary sequestration showing an extensive right-sided echogenic lung mass and also normal lung tissue and normal left lung and cardiac axis (main differential diagnosis would be cystic congenital adenomatoid malformation of the lung).
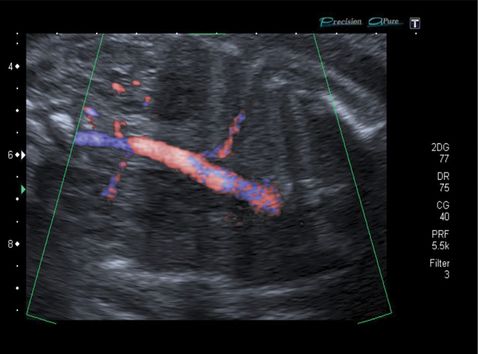
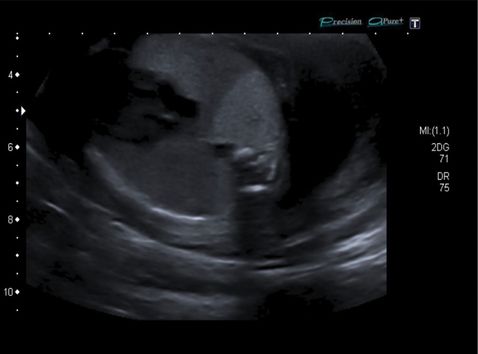
Longitudinal coronal ultrasound section with color Doppler imaging of intralobar bronchopulmonary sequestration showing typical features of an abnormal feeding vessel from a direct aortic blood supply.
Antenatal diagnosis and initial management
Sequestrations usually present at the anomaly scan with an echogenic lung mass with or without cardiac axis deviation or mediastinal shift. The main distinguishing diagnostic feature is the presence of a feeding blood vessel, which can be seen on color Doppler[12].
Referral should be made to a tertiary fetal medicine unit for a detailed anatomic survey, including an assessment of fetal anatomy, amniotic fluid index and exclusion of hydrops and a fetal echo. Karyotyping should be offered. Counseling should be offered by a pediatric surgeon and/or a neonatologist regarding the neonatal and long-term implications, and neonatal and surgical management. Termination of pregnancy would also be an option, particularly for larger or complex nonisolated lesions, and should also be discussed. Color or power Doppler may aid in identifying a feeding vessel and its source. The area under the diaphragm should be carefully inspected for abdominal components. Fetal MRI may be considered. Other differential diagnoses include CDH, CCAML, bronchogenic cyst, mediastinal hamartoma or neuroblastoma.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


