Fetal Therapy
The first open fetal surgical procedure was performed at the University of California, San Francisco (UCSF) in 1982.1 Since then, more than 515 fetal interventions have been performed at UCSF over the past 30 years without maternal mortality. Over the past 30 years, a number of advances have been developed to allow a broader application of fetal intervention. These techniques, including maternal hysterotomy, minimal-access fetoscopy, and percutaneous fetal access, were initially tested and validated in animal models. More recently, in utero therapy has shown to be beneficial for non-life-threatening conditions such as myelomeningocele (MMC). In this chapter, we will present an overview of the current state of fetal surgery and will review specific fetal problems outlining current management strategies.
General Principles
The primary morbidity following fetal surgery has been, and remains, preterm labor resulting in premature delivery, usually between 25 and 35 gestational weeks. Preventing preterm labor after fetal intervention remains problematic. Complications can also arise from endotracheal intubation, general anesthesia, epidural and spinal anesthesia, blood transfusion, premature rupture of membranes, chorioamniotic separation, chorioamnionitis, and placental abruption. Long-term morbidity from the hysterotomy includes infertility, uterine rupture with the current and future pregnancies, and mandatory cesarean section with future pregnancies. Notably, in reviewing our experience, subsequent fertility following fetal intervention has been good.2
Fetal Access
Ultrasound-guided percutaneous procedures are performed through small skin incisions on the mother’s abdominal wall. During these operations, real-time ultrasound is needed to visualize the fetal and maternal anatomy.3 Catheters and shunts can be inserted into the fetus to drain cystic masses, ascites, or pleural fluid into the amniotic space. In addition, radio frequency ablation (RFA) probes can be deployed into the amniotic space to treat various twin gestational anomalies. The needles used to place these catheters, as well as the RFA device, are approximately 1.5–2 mm in diameter, minimizing morbidity to the mother and irritation of the uterus.4,5
Open fetal procedures require general anesthesia with a combination of preoperative indomethacin and high mean alveolar concentration of inhalational agents to maintain uterine relaxation.6–8 An epidural is also inserted for postoperative analgesia.
Anomalies Amenable to Fetal Surgery
Congenital Diaphragmatic Hernia
Despite significant advances in neonatal respiratory support, survival for children born with congenital diaphragmatic hernia (CDH) remains only 60–70% throughout the USA. Additionally, survival for prenatally diagnosed CDH may be as low as 25% due to IUFD and stillborns that are not included in conventional postnatal survival data.9–11 This high mortality rate has made CDH a primary area of interest for the development of effective prenatal intervention. In fact, improving outcomes specifically for CDH was a significant driving force in the genesis of fetal surgery at UCSF.
Prognostic Criteria
One of the key elements in developing fetal intervention for CDH has been identifying what factors will identify those fetuses at the greatest risk for a poor outcome. The factors most consistently associated with a poor outcome on prenatal ultrasound are (1) the presence of liver herniation into the chest; and (2) a low lung-to-head ratio (LHR). In our experience, survival has been 100% in fetuses with CDH that do not have liver herniation on prenatal ultrasound and 56% in fetuses with CDH and liver herniation into the chest.12 The LHR is calculated as the area of the contralateral lung at the level of the cardiac atria divided by the head circumference. This LHR value has been shown to statistically correlate with survival: 100% survival with an LHR greater than 1.35, 61% survival with an LHR between 0.6 and 1.35, and 0% survival with an LHR less than 0.6.12
While the LHR has been a reliable predictor of outcomes at our center, other institutions have suggested the LHR does not account for discrepant growth rates between the head and lung during gestation and therefore may not be reliable at certain gestational ages.13,14 To account for this, the observed to expected LHR (OE LHR) has been proposed. The OE LHR is represented as a percentage of what the expected LHR would be in a normal fetus of the same gestational age. For left-sided defects, an OE LHR <25% is associated with an 18% survival whereas an OE LHR >45% correlates with 89% survival.13,15
Magnetic resonance imaging (MRI) for volumetric measurement of the lungs is a promising modality for prognosis with CDH.16 MRI can be used to calculate the percent-predicted lung volume (PPLV). Results for PPLV have varied. In one study, a PPLV >20% was associated with 100% survival whereas survival was only 40% when PPLV was <15%.17 In another study, a PPLV <25% was associated with a 13% survival and a PPLV >35% correlated with 83% survival.18 MRI can also be used to determine the percentage of liver herniation, although the prognostic value of this finding is still being investigated.18
Fetal Interventions
CDH and its effect on fetal lung development has been studied in animal models.19,20 In the fetal lamb model, compression of the lungs, either with an intrathoracic balloon or by creation of a diaphragmatic hernia, results in uniformly fatal pulmonary hypoplasia. However, in utero correction of the compressing lesion leads to sufficient lung growth and development, which improves postnatal survival.20
This concept of early, in utero correction of CDH has been studied and applied in humans.21,22 Fetal surgery for CDH initially involved open repair of the diaphragmatic defect. The first successful case was reported in 1990 which demonstrated the feasibility of open fetal repair using a two-step approach which involved creation of an abdominal silo to accommodate the reduced viscera and prevent compression of the umbilical vessels.23 This initial success was followed by a prospective trial at UCSF comparing open fetal surgery to postnatal repair in severe cases of prenatally diagnosed CDH. However, in this study, there was no difference in survival or in the need for extracorporeal membranous oxygenation (ECMO) between fetal repair and postnatal repair.22,24 Concordant with this effort, investigators at UCSF observed that fetuses with congenital high airway obstruction syndrome (CHAOS) had pulmonary hyperplasia.25 Also, fetal tracheal occlusion had been shown to cause pulmonary hyperplasia.26 In this condition, the lung parenchyma creates fluid that is ‘exhaled’ by the fetus. Occluding the trachea causes a build-up of this fluid and subsequent pulmonary hyperplasia.27,28 The inability to improve outcomes with open fetal repair for severe cases of CDH led to an interest in this physiologic process.29
The first eight patients were treated with open hysterotomy and tracheal occlusion with a metallic clip.30 This approach proved to be problematic for several reasons. First, the open hysterotomy led to significant prematurity due to premature labor. Second, the use of clips was associated with tracheal stenosis and also required a stringent delivery plan—which was later described as the ex utero intrapartum treatment (EXIT) procedure—whereby the fetus was exposed through a hysterotomy and maintained on utero–placental circulation while the clip was removed and a patent airway established prior to delivering the baby.31 However, outcomes with this approach were poor with only a 15% survival rate.30
Ongoing advancements in fetal surgery led to fetoscopic balloon placement for tracheal occlusion (Fig. 10-1). This technique has the advantages of being less invasive, a lower risk of tracheal stenosis, and the balloon being much easier to remove, although still necessitating an EXIT procedure. Results in the first eight cases were favorable with a 75% survival rate compared to a 38% survival rate in historical, case-matched controls managed with postnatal repair.32
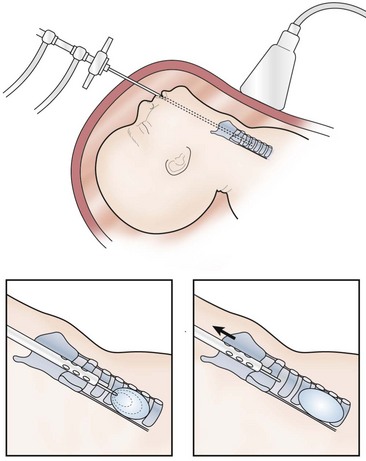
FIGURE 10-1 This schematic diagram shows the method of fetoscopic tracheal occlusion. A fetoscope is placed into the fetal mouth, the airway is identified, and a balloon is inserted into the trachea by using both fetoscopic and ultrasonographic visualization.
These early results led to an National Institutes of Health (NIH) funded, prospective randomized trial comparing in utero fetoscopic tracheal occlusion to standard postnatal care for fetuses diagnosed with severe left-sided CDH (liver up and LHR <1.4) and no other detectable anomalies. However, results of the trial showed no difference in survival between the tracheal occlusion group and the standard postnatal care group (73% vs 77%, respectively).33 Unexpectedly, the survival in the postnatal repair group was considerably greater when compared to historical controls. Although this study did not demonstrate a difference in survival between the prenatal intervention group and the postnatal group, the results of this trial demonstrate the tremendous importance of proper randomized controlled trials for novel fetal surgical procedures.
Further data regarding fetal tracheal occlusion have suggested that temporary, short-term reversible tracheal occlusion may be preferable to a longer duration of occlusion. Animal models of fetal tracheal occlusion have demonstrated that long-term tracheal occlusion can be deleterious to type II pneumocytes (the cells that secrete surfactant) and that this adverse effect is not seen with a shorter duration of tracheal occlusion.34 To test the hypothesis that temporary fetal tracheal occlusion is better, Deprest et al. studied patients undergoing fetal tracheal balloon occlusion who also had the balloon removed prenatally to limit the duration of occlusion.35 In this group of patients, improved lung growth was evident on fetal MRI and was also associated with improved postnatal survival. While reversal of the tracheal occlusion requires a second maternal and fetal intervention for balloon removal, it obviates the need for an EXIT procedure at birth. Early results have been favorable.31,36
These promising findings with temporary tracheal occlusion have led to its current application in Europe. The European FETO consortium has reported a 48% survival rate among 210 cases of severe CDH treated with temporary fetal tracheal occlusion, and the Eurofetus group is currently sponsoring a prospective fetal tracheal occlusion trial that seeks to determine the ideal time and duration for tracheal occlusion.37 Our group at UCSF is currently offering reversible, fetal tracheal balloon occlusion for fetuses with liver herniation in the chest and an LHR of <1.0, as these babies continue to have a very high mortality.38 This study has Food and Drug Administration oversight, and involves percutaneous placement of a fetoscopic tracheal balloon between 26 and 28 weeks gestation, with removal of the balloon via a second percutaneous fetoscopic procedure between 32 and 34 weeks.
Neoplasms
Fortunately, fetal neoplasms are rare. When they do occur, most are benign. However, if they become large enough, they can impede venous return to the heart or cause high-output heart failure via arteriovenous shunting. Such shunting can lead to non-immune fetal hydropic changes such as polyhydramnios, placentomegaly, skin and scalp edema, and pleural, pericardial, and peritoneal fluid accumulation. When only one compartment is involved, this is considered early fetal hydrops; when two or more compartments are affected, then true hydrops is present. If left untreated, hydrops is nearly always fatal.39–40 The two most common prenatally diagnosed neoplasms that cause nonimmune fetal hydrops are congenital pulmonary airway malformations (CPAM) and sacrococcygeal teratomas (SCT).
Congenital Pulmonary Airway Malformations
CPAMs are pulmonary lesions with a broad range of clinical presentations. This new terminology includes congenital cystic adenomatoid malformations (CCAM) and bronchopulmonary sequestrations. CCAMs are much more likely than sequestrations to cause nonimmune fetal hydrops. CCAMs are characterized by an overgrowth of respiratory bronchioles with the formation of cysts of various sizes.41–44 Most fetuses diagnosed with a CCAM develop normally, and can be followed with serial ultrasound studies. These asymptomatic patients then undergo standard, postnatal resection. A small percentage of patients with the prenatal diagnosis of CCAM will develop non-immune hydrops.43,44
Various measurements have been developed to predict which fetuses are at risk for developing hydrops. The most accepted measurement is the CCAM volume ratio (CVR), defined as the product of the three longest measurements of the lesion on ultrasound multiplied by the constant 0.52, and then divided by the head circumference. Crombleholme and colleagues identified a CVR of 1.6 as a cut-off for an increased likelihood of developing hydrops.45 When the CVR is <1.6, there is only a 2% risk of developing hydrops. When the CVR is >1.6, there is an 80% chance of developing hydrops.
CCAMs that are predominantly microcystic have a more predictable course than the macrocystic ones. Microcystic or solid CCAMs undergo steady growth that tends to plateau at 26 to 28 weeks gestation. At this point, fetal growth exceeds that of the CCAM. For this reason, patients with microcystic or solid CCAMs should be followed closely up to 26 to 28 weeks gestation at which point the interval between ultrasound examinations can be lengthened if the pregnancy has been otherwise uncomplicated. In contrast, macrocystic CCAMs undergo abrupt enlargement due to rapid fluid accumulation in a dominant cyst. Therefore, macrocystic CCAMs require close follow-up with serial ultrasound throughout the duration of the pregnancy.42,46
If a fetus develops hydrops at a viable gestational age, early delivery should be considered. Hydropic fetuses who are not yet viable outside the uterus, and have a dominant macrocystic lesion, are appropriate candidates for a thoracoamniotic shunt.47 Needle drainage alone has not been found to be an effective therapy as rapid re-accumulation of fluid in the cyst necessitates repeat intervention. In the largest single-center experience with thoracoamniotic shunts, shunting led to a mean 51% volume reduction in the size of the lesion and a 70% survival rate.48 Other institutions have reported similar survival rates.49 Despite shunting, these babies can still have significant respiratory distress at birth and should be delivered at a tertiary referral center.
Open fetal thoracotomy and CCAM resection is an option in the pre-viable fetus with a microcystic or solid lesion. This is performed through an open hysterotomy. A thoracotomy is made through the fifth intercostal space, and the lobe containing the CCAM is identified and exteriorized through the incision (Fig. 10-2). The pulmonary hilar structures are then mass ligated using an endoloop or endoscopic stapler. The thoracotomy is then closed in layers.50–51
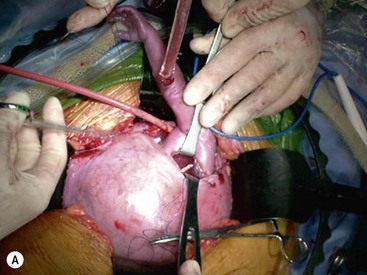
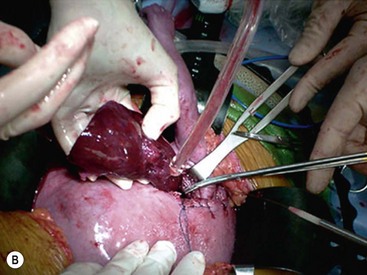
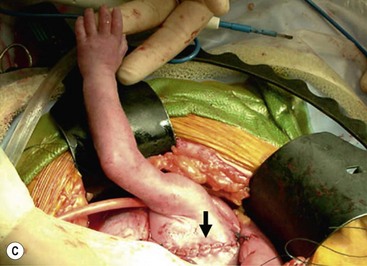
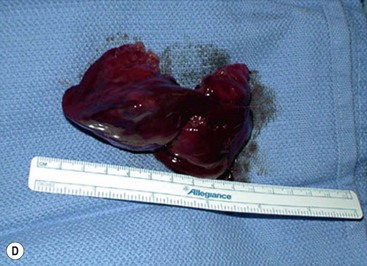
FIGURE 10-2 These photographs depict an infant with a large left upper lobe CCAM undergoing in utero left lobectomy. (A) The infant’s left arm is visualized. Note the maternal hysterotomy and the left fetal thoracotomy (with retractors inserted) through the fifth intercostal space. (B) The left upper lobe containing the CCAM has been identified and exteriorized through the thoracotomy incision. The pulmonary hilar structures were mass ligated using an endoloop. (C) The fetal thoracotomy incision (arrow) has been closed. (D) The left upper lobe specimen containing the CCAM is seen.
In a group of 120 patients with the prenatal diagnosis of CPAM from UCSF and Children’s Hospital of Philadelphia (CHOP), 79 had no evidence of hydrops.51 Of these, 76 were followed expectantly and all survived. Three fetuses without evidence of hydrops and with large dominant cysts underwent thoracoamniotic shunting. All three fetuses survived. Twenty-five hydropic fetuses were followed with no intervention. All mothers delivered prematurely and all fetuses died perinatally. Sixteen fetuses with hydrops underwent intervention: 13 underwent open fetal surgery while three underwent thoracoamniotic shunting. Two of the three survived in the group that underwent shunt insertion and eight of 13 survived in the open fetal surgery group.
Despite positive results with open fetal resection in the hydropic fetus, there has been a shift away from this therapy in the last five years due to the efficacy of maternal steroids. This finding was discovered serendipitously at UCSF during the preparation of several hydropic fetuses for open fetal surgery.52 In these cases, maternal steroids were administered to enhance fetal lung maturity. Preoperative ultrasound studies showed resolution of the hydrops and those fetuses survived to delivery and beyond. Thirteen patients with microcystic CCAMs, nine of which were complicated by hydrops, had an overall survival rate of 85% with resolution of hydrops in seven of nine fetuses.53 CHOP has reported a series of 11 patients, five of which had hydrops, and all survived after receiving steroids.54
Currently, we recommend maternal betamethasone for fetuses with nonimmune hydrops or a CVR >1.6. Steroids can be re-dosed, but repeated administration of maternal steroids beyond three to five courses can result in untoward effects such as reduced birth weight.55 It is widely accepted that steroids are most effective for predominantly microcystic or solid lesions as this is the component of the malformation that responds to steroids. Macrocystic lesions are less likely to respond.



