Chapter 10
Fetal and Neonatal Neurologic Injury
Tania F. Esakoff MD, Sarah J. Kilpatrick MD, PhD, Arvind Palanisamy MBBS, MD, FRCA
Chapter Outline
The detection and diagnosis of fetal and neonatal brain injury have been advanced by improvements in functional imaging and the identification of potential biochemical markers. Evidence indicates that inflammatory mediators play an important role in the pathophysiology of fetal brain injury. Evidence also suggests that maternal administration of magnesium sulfate before anticipated early preterm birth reduces the risk for cerebral palsy in surviving infants. New data suggest that induced hypothermia is beneficial for the treatment of neonatal hypoxic ischemic encephalopathy. Of specific concern to anesthesia providers is rodent and nonhuman primate data that suggest that fetal exposure to anesthetic agents may have harmful effects on neurogenesis and synapse formation in the developing brain. Overall, however, little progress has been made in reducing the incidence of neonatal brain injury and cerebral palsy.
Fetal Brain Development
Generation of the various cell types that populate the developing brain, and the subsequent layering and organization, is a precisely regulated process encoded by genetic programs and modified by epigenetic influences.1–4 Contrary to previous dogma, it is now well established that the brain continuously evolves during ontogeny and that these processes are susceptible to subtle changes in the internal and external milieu. Although such neurodevelopmental processes occur throughout the human lifespan, the process is most robust and dynamic during the perinatal period.5 Much of our understanding of the processes that drive fetal brain development comes from studies in rodents and nonhuman primates.6 With recent advances in neuroimaging, it is now possible to study brain anatomy and assess neurobehavioral changes in the human fetus.7
When pathways leading to orderly brain development are deconstructed, three major events appear critical to the establishment of functional synapses. Neuronal proliferation, migration, and cellular differentiation occur in a preordained fashion to establish early neural circuitry. These processes often overlap and occur at different rates in different brain regions. Neurogenesis, a term that encompasses both neuronal proliferation and subsequent survival, begins with neural stem/progenitor cells in neurogenic niches such as the subventricular zone and the subgranular zone of the dentate gyrus. These neural progenitor cells undergo mitosis to generate immature neurons that migrate in a radial fashion and laminate the cortex in an “inside-out” fashion.8 Interneurons, which comprise 10% to 15% of the total neuronal cells in the brain, originate from the ganglionic eminences in the developing brain.9 These newly generated interneurons, which play an indispensable role in circuit inhibition, migrate in a tangential manner to populate distinct brain areas. Both forms of migration are guided by cell-intrinsic mechanisms as well as by structural scaffolds and humoral mediators such as gamma-aminobutyric acid (GABA) and glutamate.10,11
In humans, neurogenesis starts and peaks at 5 and 25 weeks’ gestation, respectively, while neuronal migration is completed between 30 and 36 weeks’ gestation.12 Between 20 and 40 weeks’ gestation, these processes are followed by the generation of an array of supporting glial cells, such as astrocytes and oligodendrocytes.12 Concurrently, synapse formation begins as early as the 10th week of gestation and continues to increase gradually at a rate of approximately 4% per week until the end of the second trimester. After this phase, a robust and exponential increase in synapse formation (almost 40,000 synapses/min) occurs between 28 weeks’ and term gestation.13 These processes, in conjunction with the onset of myelination, result in a fivefold increase in brain volume and the appearance of morphologic features of the mature brain such as sulci and gyri. By 24 weeks’ gestation the fetus has all the neural machinery necessary to perceive pain.14 Many clinicians recommend that appropriate measures should be taken to provide fetal analgesia during fetal surgical procedures from this point onward.15
Although the ontogeny of neurotransmitter systems is less well studied, a wealth of animal and human data indicates that these systems appear very early in life, before the phase of active synaptogenesis.12 The presence of these neuromodulatory substances before synapse formation lends credence to the view that they serve a trophic role during early brain development, a role that is distinct from their predominant role of facilitating synaptic neurotransmission in the mature brain. Among these neurotransmitters, GABA remains the most widely studied (Figure 10-1).16 Although GABA has an inhibitory action in the mature brain, GABA serves an excitatory role during fetal brain development. The major mechanism for this role reversal is the differential expression of chloride ion transporters NKCC1 and KCC2; these transporters increase the intracellular concentration of chloride in developing neurons.17 On stimulation of GABA receptors that are expressed in neural progenitor cells and immature neurons, chloride ions are actively extruded, causing membrane depolarization rather than the hyperpolarization seen in mature neurons. This depolarizing effect of GABA decreases DNA synthesis and inhibits proliferation of neural progenitor cells,18 causes concentration- and time-dependent effects on neuronal migration,11 and plays a major role in activity-dependent synapse formation.19
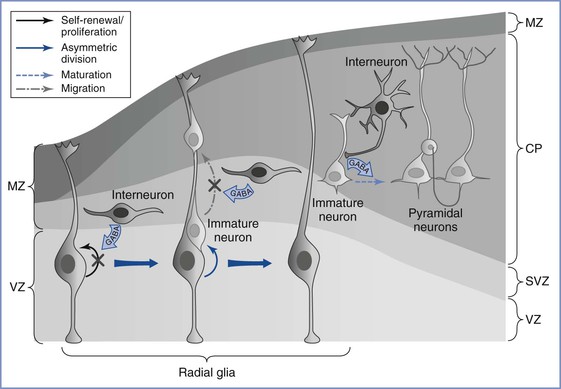
FIGURE 10-1 Gamma–aminobutyric acid’s (GABA) role in regulating embryonic cortical development. During corticogenesis, interneurons migrating in the subventricular zone (SVZ) can release GABA and activate GABAergic receptors on the radial glia, depolarizing these progenitors and decreasing their proliferation. Radial glia generate immature pyramidal neurons through asymmetric division, and the migration of these immature neurons along the radial fibers is decreased by GABA signaling. As young neurons assume their position in the cortex and begin to mature, GABA-mediated depolarization by the interneurons is required for the development of dendritic arbors and excitatory synaptic inputs from other pyramidal neurons. MZ, marginal zone; VZ, ventricular zone; SVZ, subventricular zone; CP, cortical plate. (From Wang DD, Kriegstein AR. Defining the role of GABA in cortical development. J Physiol 2009; 587:1873-9.)
The N-methyl-D-aspartate (NMDA)-subtype glutamate receptors originate later than the GABA receptors and remain functionally silent because of magnesium ion–induced channel blockade; thus, they play a limited role during early brain development. Dopaminergic, cholinergic, and serotonergic systems develop concomitantly and appear fully functional by the second trimester.12 Pharmacologic interventions (e.g., ethanol, antiepileptic drugs) that act directly or indirectly on these powerful neuromodulator systems induce long-lasting impairment of fetal brain development, mainly owing to impaired neurogenesis and/or altered neuronal migration.20–22 Alteration of this excitation-inhibition balance is purported to be responsible for an array of childhood neurodevelopmental disorders.
Experimental studies reveal that the fetal blood-brain barrier is morphologically well developed and functionally competent at term.23 Convincing evidence confirms that the endothelial tight junctions of the blood-brain barrier are as effective in the term fetus as in the adult, although the exact time that blood-brain barrier competency is established in the human fetus is unknown. In rodents, data suggest that the fetal blood-brain barrier is established between embryonic days 11 and 17 (term gestation is 22 days), a time period that corresponds to approximately the late second and early third trimesters in humans.24
Cerebral Palsy
History, Definitions, and Significance
In 1861, John Little, an orthopedic surgeon, first described cerebral palsy in a report to the Obstetrical Society of London. Described as a neonatal neurologic disorder associated with difficult labor or birth trauma, the disorder was known as Little’s disease until William Osler coined the term cerebral palsy in 1888.25 A precise definition and classification of cerebral palsy has proved elusive. In the forward to the “Report on the Definition and Classification of Cerebral Palsy,” published in 2007 in Developmental Medicine and Child Neurology, Peter Baxter25 wrote, “This [supplement] illustrates the difficulties inherent in trying to agree what we mean by the terms we use and that a classification that suits one purpose, such as a diagnostic approach, may not always be ideal for others, such as therapy issues. Defining and classifying cerebral palsy is far from easy. We do need a consensus that can be used in all aspects of day-to-day care and for future research on cerebral palsy.”
Today, cerebral palsy is defined as a nonprogressive disorder of the central nervous system (CNS) present since birth that includes some impairment of motor function or posture.25 Intellectual disability (formerly known as mental retardation) may be present but is not an essential diagnostic criterion. Various forms of cerebral palsy exist, with differences in pathology, pathophysiology, and potential relationships with intrapartum events. The literature on cerebral palsy is difficult to review and understand. Data from individual studies are difficult to compare because of variations in the duration of follow-up, birth weight classifications, inclusion criteria for congenital abnormalities, and exclusion criteria for various causes of death. Terms such as hypoxic-ischemic encephalopathy of the newborn, newborn asphyxia, birth asphyxia, and asphyxia neonatorum are difficult to distinguish. Some authorities, including the American College of Obstetricians and Gynecologists (ACOG), have argued that the term birth asphyxia should be abandoned.26
Intrapartum events continue to receive the blame for some cases of cerebral palsy. It is a widely believed theory that an intrapartum reduction in fetal oxygen delivery may cause cerebral palsy, and early reports in primates demonstrated that perinatal asphyxia could cause brain injury.27 Continuous electronic fetal heart rate (FHR) monitoring, which has largely replaced intermittent FHR auscultation during labor, is believed to prompt the delivery of at-risk fetuses and thus reduce asphyxial events. However, despite a higher incidence of cesarean delivery, no reduction in the incidence of cerebral palsy has been observed since the widespread implementation of continuous electronic FHR monitoring during labor.28,29 Further, among patients with new-onset late FHR decelerations, an estimated 99% of tracings would be false positive “if used as an indicator for subsequent development of cerebral palsy.”30 This information is probably surprising to the lay public and trial lawyers as well as to obstetricians; a survey of maternal-fetal medicine fellows showed that they, too, have greatly overestimated the diagnostic accuracy of FHR monitoring.30
Large randomized trials have not demonstrated better fetal and neonatal outcomes with continuous electronic FHR monitoring than with intermittent FHR auscultation.31,32 In an editorial citing observations made by Schifrin and Dame,33 Friedman34 opined, “The absence of either suggestive or overtly ominous fetal heart rate patterns is reliably reassuring.” Unfortunately, there is little objective evidence that reassuring FHR tracings exclude the subsequent occurrence of cerebral palsy. In a 1993 review of published FHR monitoring studies, Rosen and Dickinson35 could not identify FHR patterns that were consistently associated with neurologic injuries. Moreover, no consistent FHR pattern was observed in a subset of 55 brain-damaged infants. These investigators concluded, “We do not advocate the abandonment of the use of electronic fetal monitoring, but we do believe that it is yet to be proved to be of value in predicting or preventing neurologic morbidity.” A more focused application of FHR monitoring may ultimately be found useful. For example, fetal inflammatory changes, which can be associated with neurologic injury, may be associated with characteristic FHR findings.36 Thus, electronic FHR monitoring provides incomplete data that should be evaluated in the clinical context in which is it used.
Despite significant limitations in the use of intrapartum electronic FHR monitoring, there is no doubt that it will continue to be used for the foreseeable future. In a review of medicolegal issues in FHR monitoring, Schifrin and Cohen37 noted that despite its limitations, “Monitoring deserves credit for reducing intrapartum death, one of the original rationales for its development.”
A 2008 workshop (sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the ACOG, and the Society for Maternal-Fetal Medicine [SMFM]) updated the definitions of various types of FHR tracings to simplify interpretation for providers.38 As a result, a three-category system was developed. Category I FHR tracings provide the most reassuring evidence of fetal well-being and strongly predict normal fetal acid-base status at the time of observation. Category III tracings are the most ominous; these tracings predict abnormal fetal acid-base status at the time of observation and require prompt evaluation. Most FHR tracings are category II (indeterminate). Category II tracings are not predictive of abnormal fetal acid-base status, but they do not provide sufficient evidence to be classified as either normal or abnormal; these tracings require continued surveillance and reevaluation (see Chapter 8).
Intrapartum events are responsible for some cases of cerebral palsy39; however, these cases are few. After exclusion of infants with significant congenital anomalies, intrapartum events—including asphyxial insults—likely account for only 5% to 8% of all cases of cerebral palsy at all gestational ages.40,41 In 1999, the International Task Force on Cerebral Palsy published a consensus statement summarizing criteria that are necessary and suggestive of an intrapartum etiology for neurologic abnormalities (Box 10-1).42 In 2010, a proposed evidence-based neonatal workup to confirm or refute allegations of intrapartum asphyxia was published.43
Epidemiology and Etiology
The causes of cerebral palsy are not known, but the varying forms suggest a multifactorial etiology. The Collaborative Perinatal Project still represents one of the largest studies of the antecedent factors associated with cerebral palsy. The investigators in this study evaluated the outcomes of 54,000 pregnancies among patients who delivered at 12 university hospitals between 1959 and 1966. They evaluated more than 400 variables in a univariate analysis,44 which identified potential risk factors that were then subjected to a more rigorous multivariate analysis.45 Maternal age, parity, socioeconomic status, smoking history, maternal diabetes, duration of labor, or use of anesthesia was not associated with cerebral palsy in the univariate analysis. The factors most strongly associated with cerebral palsy in the multivariate analysis were (1) maternal mental retardation, (2) birth weight of 2000 g or less, and (3) fetal malformations. Other factors associated with cerebral palsy included (1) breech presentation (but not vaginal breech delivery), (2) severe proteinuria (more than 5 g/24 h) during the second half of pregnancy, (3) third-trimester bleeding, and (4) a gestational age of 32 weeks or less. There was a slight association between cerebral palsy and fetal bradycardia, chorioamnionitis, and low placental weight. However, only 37% of the cases of cerebral palsy occurred in patients with one or more of these identified risk factors.
Rosen and Dickinson46 reviewed studies from Europe, Australia, and the United States that were published between 1985 and 1990 and included data from 1959 to 1982. The incidence of cerebral palsy ranged from 1.8 to 4.9 (composite rate of 2.7) cases per 1000 live births. The incidence of certain conditions in infants with cerebral palsy was as follows: birth weight less than 2500 g, 26%; diplegia, 34%; hemiplegia, 30%; quadriplegia, 20%; and extrapyramidal forms, 16%.
Two more recent studies from Australia reexamined the risk factors for cerebral palsy. A large epidemiologic study from 1998 noted an incidence of neonatal encephalopathy of 3.8 per 1000 term births.47 The investigators identified preconception and antepartum factors that were associated with neonatal encephalopathy (Box 10-2). In the second study from 2011, the greatest risks for cerebral palsy included (1) preterm birth, (2) fetal growth restriction (also known as intrauterine growth restriction), (3) perinatal infection, and (4) multiple gestation.48 Upper respiratory tract and gastrointestinal infections during pregnancy and instrumental (forceps or vacuum) vaginal delivery were not associated with cerebral palsy.48 Evidence suggests that intrapartum factors alone are associated with neonatal encephalopathy in less than 5% of cases.47,49 These data, along with the recognition that most patients with identified risk factors do not have children with cerebral palsy, have led the majority of investigators to agree that most cases of cerebral palsy cannot be predicted and that the identification of pregnancy-related conditions contributes minimally to the identification of patients at risk for having a child with cerebral palsy.
In 2000, the ACOG and the American Academy of Pediatrics (AAP) convened the Neonatal Encephalopathy and Cerebral Palsy Task Force. The resulting landmark report,49 which was released in 2003, was reviewed and endorsed by such groups as the U.S. Department of Health, the National Institutes of Health, the SMFM, the Child Neurology Society, the March of Dimes Birth Defects Foundation, the Society of Obstetricians and Gynaecologists of Canada, and the Royal Australian and New Zealand College of Obstetricians and Gynaecologists. The Task Force extended the earlier international consensus statement regarding the requirements for establishing a causal relationship between intrapartum events and cerebral palsy (see Box 10-1).42 The consensus statement led to several medicolegal conclusions42,49:
Phelan et al.50 subsequently confirmed that fetuses that experienced a sudden and sustained deterioration of the FHR, and that subsequently were found to have cerebral palsy, demonstrated characteristics consistent with the ACOG/AAP Task Force criteria50 for intrapartum asphyxial injury.
Peripartum Asphyxia and Cerebral Palsy
Asphyxia may be defined as insufficient exchange of respiratory gases.51 Although accurate, this definition does not include an index of severity or have any predictive value. Unfortunately, most studies have not used a uniform definition of birth asphyxia.52–54
In 1953, Dr. Virginia Apgar, an anesthesiologist, introduced her scoring system to identify newborn infants in need of resuscitation and to assess the adequacy of subsequent resuscitation efforts.55 Although the Apgar score has also been used to identify infants at risk for cerebral palsy, only a weak association has been found.56,57 In the Collaborative Perinatal Project, only 1.7% of children with a 1-minute Apgar score of 3 or less developed cerebral palsy.58 Among infants who weighed more than 2500 g at delivery, the incidence of cerebral palsy was 4.7% if the 5-minute Apgar score was 0 to 3 and 0.2% if the 5-minute Apgar score was at least 7. Among infants who weighed less than 2500 g with the same 5-minute Apgar scores, the incidence of cerebral palsy was 6.7% and 0.8%, respectively. Among all infants, a higher incidence of cerebral palsy was observed if the Apgar score remained 3 or less for longer than 5 minutes. The incidence of early neonatal death increased among those infants with prolonged neonatal depression.
Most infants who subsequently manifest evidence of cerebral palsy have a normal 5-minute Apgar score. In the Collaborative Perinatal Project, only 15% of the infants in whom cerebral palsy later developed had a 5-minute Apgar score of 3 or less, approximately 12% had a score of 4 to 6, and the remaining 73% had a score of at least 7.58 It must also be noted that preterm delivery is independently associated with a low Apgar score.
Although most cases of cerebral palsy are not attributed to intrapartum insults, intrapartum asphyxia does occur and can have serious consequences. However, the degree of asphyxia necessary to produce irreversible CNS injury is unclear. In some cases, an intrapartum insult that might have otherwise been innocuous might be superimposed on subclinical chronic fetal compromise and result in permanent injury.
Umbilical cord blood gas measurements are often used to diagnose suspected asphyxia. However, the definition of normal umbilical cord blood gas and pH measurements remains unclear.51 In one study of 15,073 vigorous neonates (arbitrarily defined as having a 5-minute Apgar score of 7 or more) conducted between 1977 and 1993, the median umbilical arterial blood gas measurements (with the 2.5th percentile in parentheses) were as follows: pH 7.26 (7.10), PO2 17 (6) mm Hg, PCO2 52 (74) mm Hg, and base excess −4 (−11) mEq/L.51 Only small differences in median pH and other measurements were present when infants were grouped according to gestational age. These data suggest that umbilical arterial blood pH in vigorous neonates can be as low as 7.10 and base excess may be as low as −11 mEq/L.
Although intrapartum events are most likely associated with a minority of cerebral palsy cases, clinical studies have attempted to define the associated extent and duration of perinatal asphyxia. Fee et al.59 defined asphyxia as an umbilical arterial blood pH of less than 7.05 with a base deficit greater than 10 mEq/L; they concluded that this threshold was a poor predictor of adverse neurologic outcomes. Goodwin et al.60 defined asphyxia as an umbilical arterial blood pH of less than 7.00; with the use of this definition, hypoxic ischemic encephalopathy and abnormal neurologic outcome were associated with acidemia. Goldaber et al.61 also observed greater neonatal morbidity and mortality among term infants (birth weight > 2500 g) with an umbilical arterial blood pH of less than 7.00.
Low et al.62,63 also studied complications of intrapartum asphyxia in term and preterm infants. They developed a complication score that expressed the magnitude of neonatal complications. Among term infants, the frequency and severity of newborn complications increased with the severity and duration of metabolic acidosis at birth. Importantly, respiratory acidosis at birth did not predict complications in newborns. Similar results were noted for preterm infants delivered between 32 and 36 weeks’ gestation. In contrast, in infants delivered before 32 weeks’ gestation, complications were similar in the control and asphyxia (defined as umbilical arterial blood buffer base < 30 mmol/L) groups. When this scoring system was used in term infants, the threshold for moderate or severe newborn complications was an umbilical arterial blood base deficit of 12 mmol/L.63
Relatively few studies have followed neurodevelopmental examinations for a sufficient duration to make meaningful conclusions about peripartum predictors of neurologic injury. Nagel et al.64 performed such examinations in 30 children in whom umbilical arterial blood pH was less than 7.00 at delivery, 28 of whom survived the neonatal period. Evaluation at 1 to 3 years of age detected 3 children who had experienced an episode of hypertonia. The majority of children exhibited no major problems, with only 1 child displaying mild motor developmental delay. Another study examined neonatal complications (neonatal death, grade 3 or 4 intraventricular hemorrhage, gastrointestinal dysfunction, and neonatal seizures) in 35 newborns with an umbilical arterial blood pH of less than 7.00 at delivery, 3 of whom died during the neonatal period.65 An umbilical arterial blood base deficit greater than or equal to 16 mmol/L and a 5-minute Apgar score less than 7 had a sensitivity and specificity for predicting adverse neonatal outcomes of 79% and 81%, respectively. These investigators did not perform any follow-up neurologic examinations after the neonatal period.65
Because metabolic acidosis may be a predictor of complications in newborns, the severity of intrapartum acidosis could be an important variable. Gull et al.66 studied a small cohort of 27 patients with terminal bradycardia who were delivered vaginally. Not surprisingly, the umbilical arterial blood base deficit was greater in infants with end-stage bradycardia than in controls. The loss of short-term FHR variability for more than 4 minutes during terminal bradycardia correlated with the development of metabolic acidosis.
The relationship between umbilical arterial blood base excess values and the timing of hypoxic injury has been estimated in human and animal studies.67 In addition, in a 2010 systematic review and meta-analysis, an umbilical cord arterial blood pH less than 7.00 was significantly associated with important, biologically plausible, adverse neonatal outcomes (i.e., neonatal mortality, hypoxic ischemic encephalopathy, intraventricular hemorrhage, periventricular leukomalacia, cerebral palsy).68 Unfortunately, this relationship does not consider the role of previous or repetitive hypoxic episodes before the episode in question and therefore cannot accurately pinpoint the time of injury. Fortunately, the human fetus is quite robust, and episodes of intrauterine asphyxia usually yield a normal neonate. A much smaller number of fetuses experiencing such episodes die in utero. Blumenthal69 concluded that there is a fine threshold between normality and death from asphyxia.
The increased presence of nucleated red blood cells in the umbilical circulation at delivery has been proposed as a marker of the occurrence and timing of intrauterine asphyxia.50,70,71 However, data from these investigations demonstrated considerable variability and were influenced by birth weight and gestational age.72 Furthermore, the timing of the injury may be difficult to determine with confidence, because multiple episodes of asphyxia may have occurred. In such cases, the nucleated red blood cell count may reflect and implicate only the most recent, but possibly least important, event. Both nucleated red blood cell and lymphocyte counts appear to undergo more sustained elevations in cases of antepartum asphyxia than in cases of intrapartum asphyxia.49
Chorioamnionitis, Fever, and Cerebral Palsy
An association between cerebral palsy and chorioamnionitis has been demonstrated in preterm infants and term infants of normal birth weight.73,74 An elevated maternal temperature is one sign of chorioamnionitis, but alone it is insufficient for the diagnosis. Other signs include, but are not limited to, maternal and fetal tachycardia, foul-smelling amniotic fluid, uterine tenderness, and maternal leukocytosis. The diagnosis remains unproven until confirmed by placental culture or histologic examination.
The mechanism by which chorioamnionitis is associated with cerebral palsy is unclear; however, inflammatory cytokines may play a role (see later discussion).75–77 A landmark meta-analysis published in 2000 reported that both clinical and histologic chorioamnionitis were strongly associated with an increased risk for cerebral palsy and periventricular leukomalacia in both preterm and term infants.78 In a 2010 meta-analysis, both histologic (pooled odds ratio [OR], 1.83; 95% confidence interval [CI], 1.17 to 2.89) and clinical chorioamnionitis (OR, 2.42; 95% CI, 1.52 to 3.84) were again found to be significantly associated with cerebral palsy.79
Several studies have demonstrated a tendency for maternal temperature to rise after administration of epidural analgesia during labor (see Chapter 37).80 Although the mechanism of epidural analgesia–associated maternal pyrexia remains unclear, fever due to epidural analgesia alone is not associated with cerebral palsy.81 Epidural analgesia has been blamed for the common obstetric practice of antibiotic administration to mothers with fever but no other evidence of chorioamnionitis. This practice may lead to unnecessary neonatal sepsis evaluations and antibiotic exposure.82 Rather than treat all women with pyrexia for presumed chorioamnionitis, Mayer et al.83 correctly noted that physicians should make an effort to differentiate true chorioamnionitis from incidental maternal fever. These investigators found that additional signs of chorioamnionitis were present in all cases in which the diagnosis was later confirmed by culture or pathologic examination. Neuraxial anesthesia is not a risk factor for cerebral palsy.44
The mode of delivery has been examined as an independent risk factor for periventricular leukomalacia, the most common form of ischemic brain injury in surviving preterm infants in whom cerebral palsy later develops. In 99 women with chorioamnionitis who delivered between 25 and 32 weeks’ gestation, vaginal delivery had a significant association with periventricular leukomalacia.84 Because the cesarean delivery group likely included a substantial number of infants with nonreassuring FHR tracings, the better outcomes in these infants is striking. Prospective trials are needed to confirm these retrospective observations.
Pathophysiology of Fetal Asphyxia
Intrauterine Hypoxemia and the Fetal Brain
The fetus is exclusively dependent on the placenta for oxygen and nutrients; thus, acute and chronic conditions that affect the placenta or the umbilical cord can deprive the fetus of one or more of these vital resources. Recent evidence from experimental animal models and humans suggest that both hypoxemic and inflammatory pathways interact and augment fetal brain damage.
The spectrum of neurologic injury in neonates depends on the duration and gestational age at hypoxemic-ischemic insult. Acute hypoxemia during the early- to mid-gestational period in sheep affects the predominant neurodevelopmental events such as neurogenesis and neuronal migration. Such hypoxemia causes the death of cerebellar Purkinje cells and hippocampal pyramidal neurons, as well as impaired neuronal migration.85 In contrast, acute hypoxemia in late gestation appears to spare the hippocampus and cerebellum but causes neuronal death in the cerebral cortex and striatum.86 Furthermore, acute perinatal anoxia causes long-term changes in dendritic arborization and synaptic connectivity.87,88
Experimental models of chronic hypoxemia, based on restriction of placental mass or blood flow, demonstrate an array of completely different effects on the fetal brain. Chronic placental insufficiency relatively spares the fetal brain compared with other organ systems, although it results in reduced fetal brain weight. Overall, neurons appear to survive chronic and mild hypoxemia; even minor behavioral changes appear to resolve fully by adulthood in animal models. It is not known whether these effects are mediated by hypoxemia per se, or by other accompanying conditions such as chronic reduction of fetal nutrient supply or altered maternal-fetal endocrine status.
Dysregulation of neuronal calcium transport appears to be the initial pathway by which cerebral hypoxemia causes perinatal neuronal injury.89 Hypoxia-induced changes in the NMDA receptor increases cellular permeability to calcium; such increases in intracellular calcium trigger a variety of downstream effects, ultimately resulting in generation of free radicals, peroxidation of lipid membranes, and nuclear fragmentation. It has long been recognized that developing oligodendroglia are highly vulnerable to excitotoxic injury in preterm infants.90 Altered maturation or premature oligodendrocyte death can occur in areas of severe hypoxia-ischemia as a result of up-regulation of inflammatory cytokines by activated microglia, elevated glutamate levels, or depleted levels of the antioxidant glutathione. It is highly likely that a combination of these mechanisms, modified by the nature and duration of insults and gestational age, determine the ultimate neurobehavioral phenotype.
Maternal Inflammation and Fetal Brain Injury
Although the development of the fetal brain is encoded by genetic programming, such programs remain highly susceptible to environmentally induced epigenetic modifications and appear closely intertwined with maternal immune and endocrine systems. Recent experimental and epidemiologic studies reveal that maternal infection and inflammation early in pregnancy can cause an array of neurodevelopmental abnormalities in the offspring such as schizophrenia and autism.91–94 Among maternal infections, chorioamnionitis is the best characterized and thoroughly investigated model of perinatal neuroinflammation. Although the exact contribution of maternal inflammation to perinatal brain injury is obscured because of the association of chorioamnionitis with preterm delivery and hypoxic-ischemic encephalopathy, inflammatory experimental models have revealed much information on cytokine induction, their transport across the placenta and amniotic fluid, and subsequent activation of the fetal immune system.
The exact mechanism by which maternal inflammation triggers a fetal immune response is likely multifactorial (Figure 10-2). Despite the presence of circulating immune cells as early as 7 weeks’ gestation in humans, antigen presentation is suboptimal because of reduced expression of the major histocompatibility complex class II on antigen-presenting cells. Furthermore, the T cells are relatively immature. Therefore, maternally derived humoral mediators seem credible candidates to initiate and perpetuate an inflammatory cascade across the placenta. This idea has gained traction with the identification of maternal interleukin-6 (IL-6) in the fetal circulation as early as the second trimester, suggesting the possibility of transplacental transfer of proinflammatory cytokines.95 Proinflammatory mediators such as IL-6 cause significant impairment of placental blood flow and fetal hypoxemia in animal models, dysregulate the barrier function of both the placenta and the immature fetal blood-brain barrier, trigger production of acute-phase proteins from the fetal liver, promote T-cell entry into the immature brain parenchyma, and disrupt the orderly patterning of the fetal cerebral cortex.91,93,96–99 The role of inflammatory mediators in this phenomenon is reinforced by the direct correlation that exists between plasma levels of IL-6 and the severity of functional deficits in offspring.100,101 In addition to IL-6, cytokines such as IL-1β, IL-7, and IL-13 are up-regulated in the fetal brain after a prenatal immune insult, a phenomenon that suggests collective activation of the innate fetal immune response.102
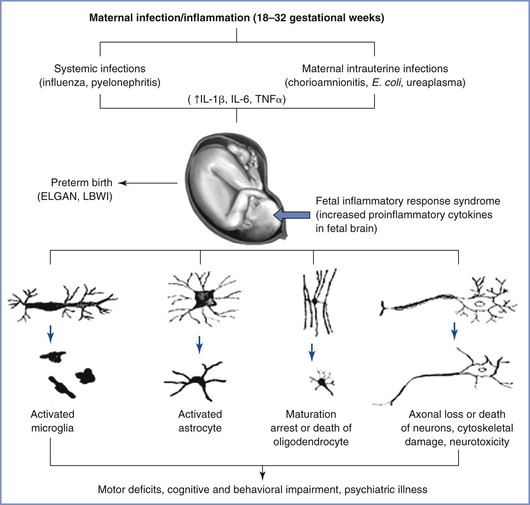
FIGURE 10-2 Probable mechanisms of fetal brain injury with in utero exposure to maternal inflammation. IL, interleukin; TNF, tissue necrosis factor; LBWI, low birth weight infant; ELGAN, extremely low gestational age neonate. (From Burd I, Balakrishnan B, Kannan S. Models of fetal brain injury, intrauterine inflammation, and preterm birth. Am J Reprod Immunol 2012; 67:287-94.)
Both microglia (the major resident macrophages in the developing brain) and the complement system have been implicated as amplifiers of this immune response. During normal fetal development, microglia invade and colonize the fetal brain during the first and second trimesters103 and are readily activated by proinflammatory mediators such as IL-1β. Activated microglial cells either cause a direct cytotoxic effect on oligodendrocytes and impair myelination or produce long-lasting alterations in neuronal-glial crosstalk, resulting in impaired synaptic function and subsequent neurodevelopmental disorders.3,91,104
At the cellular level, numerous mechanisms are involved in propagating the prenatal immune response. Collectively, robust experimental evidence suggests that prenatal inflammation alters fetal brain development at the molecular, cellular, and circuit levels. Epidemiologic studies have shown a strong correlation between maternal infection/inflammation and neurodevelopmental disorders such as schizophrenia and autism.92,105,106
Animal Models of Fetal Asphyxia
Much of our knowledge of the fetal response to insufficient exchange of respiratory gases has been gained through the use of animal models. However, the limitations of these models must be acknowledged. Raju107 reviewed the various animal models of fetal brain injury. At birth, sheep and guinea pig brains are much closer to maturity than the human brain. In this regard, rat pup and human brains are more similar to each other because they both undergo significant extrauterine development (Figure 10-3).108 Nonetheless, the importance of this distinction has been challenged. Previously, investigators relied mainly on morphologic milestones (e.g., the brain growth spurt) to compare species at different stages of development. A computerized method attempted to more accurately compare observations among 10 species (including humans) by evaluating the mathematical relationships of more than 100 developmental events and factors (e.g., evolutionary, genetic, neurochemical, neuroanatomic).109 Although all events have not been catalogued for any one species, the iterative process allows information to be added to improve the theoretic model and is freely available online.* This method is not completely understood or accepted but may explain some of the variability observed among various models of developmental brain injury.
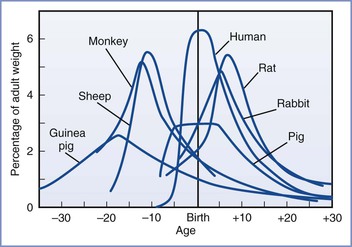
FIGURE 10-3 Brain growth spurts of seven mammalian species expressed as first-order velocity curves of the increase in weight with age. The units of time for each species are as follows: guinea pig (days); rhesus monkey (4 days); sheep (5 days); pig (weeks); human (months); rabbit (2 days); rat (days). Rates are expressed as a percentage of adult weight for each unit of time. (From Penning DH. Fetal and neonatal neurologic injury. In Chestnut DH, Polley LS, Tsen LC, Wong CA, editors. Chestnut’s Obstetric Anesthesia: Principles and Practice. 4th edition. Philadelphia, Mosby, 2009; modified from Dobbing J, Sands S. Comparative aspects of the brain growth spurt. Early Hum Dev 1979; 3:79-83.)
One advantage of the chronically instrumented fetal lamb is that it is similar in size to the human fetus, thus facilitating the placement of electrodes and vascular catheters in both the fetus and the mother. Investigators may obtain measurements while the mother (and fetus) remains anesthetized or from awake animals that have recovered from surgery. Studies of animals with continuous instrumentation allow the assessment of fetal breathing movements, gross body movements, brain electrical activity (electroencephalogram), and blood gas and pH measurements. Blood concentrations of glucose, lactate, and various hormones can also be determined. Microdialysis techniques have been used to evaluate neurotransmitter release within the fetal brain in vivo in acute, exteriorized, and chronic preparations.110–112 Other studies have measured fetal cerebral blood flow in vivo during episodes of hypoxemia113 and during maternal infusion of ethanol.114 Together, these studies have enhanced the understanding of the fetal brain response to pathophysiologic insults in utero. Ultimately, these insights may lead to improved diagnoses, treatment, and prevention of fetal brain injury.
Studies have used a variety of methods to produce fetal hypoxemia and acidemia in fetal lambs. Each method attempts to mimic one or more clinically relevant situation(s), including (1) decreased concentration of maternal inspired oxygen for several hours115 or days116; (2) decreased uterine blood flow, which may be accomplished by placement of an adjustable clamp on the common iliac artery117; (3) decreased umbilical blood flow, either by total obstruction118 or by means of a slow, progressive obstruction119; (4) selective uteroplacental embolization120; (5) maternal hemorrhage121; and (6) a combination of two insults, such as hypoxemia plus hypotension.122
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


