Chapter 599 Evaluation and Investigation
Many chromosomal loci have been identified with specific neuromuscular diseases as a result of genetic linkage studies and the isolation and cloning of a few specific genes. In some cases, such as Duchenne muscular dystrophy, the genetic defect has been shown to be a deletion of nucleotide sequences and is associated with a defective protein product, dystrophin; in other cases, such as myotonic muscular dystrophy, the genetic defect is an expansion or repetition, rather than a deletion, in a codon (a set of three consecutive nucleotide repeats that encodes for a single amino acid), with many copies of a particular codon, in this example also associated with abnormal mRNA. Some diseases manifest as autosomal dominant and autosomal recessive traits in different pedigrees; these distinct mendelian genotypes can result from different genetic mutations on different chromosomes (nemaline rod myopathy) or may be small differences in the same gene at the same chromosomal locus (myotonia congenita), despite many common phenotypic features and shared histopathologic findings in a muscle biopsy specimen. Among the several clinically defined mitochondrial myopathies, specific mtDNA deletions and tRNA point mutations are recognized. The inheritance patterns and chromosomal and mitochondrial loci of common neuromuscular diseases affecting infants and children are summarized in Table 600-1.
Clinical Manifestations
Examination of the neuromuscular system includes an assessment of muscle bulk, tone, and strength. Tone and strength should not be confused: Passive tone is range of motion around a joint; active tone is physiologic resistance to movement. Head lag when an infant is pulled to a sitting position from supine is a sign of weakness, not of low tone. Hypotonia may be associated with normal strength or with weakness; enlarged muscles may be weak or strong; thin, wasted muscles may be weak or have unexpectedly normal strength. The distribution of these components is of diagnostic importance. In general, myopathies follow a proximal distribution of weakness and muscle wasting (with the notable exception of myotonic muscular dystrophy); neuropathies are generally distal in distribution (with the notable exception of juvenile spinal muscular atrophy; Table 599-1). Involvement of the face, tongue, palate, and extraocular muscles provides an important distinction in the differential diagnosis. Tendon stretch reflexes are generally lost in neuropathies and in motor neuron diseases and are diminished but preserved in myopathies (see Table 599-1). A few specific clinical features are important in the diagnosis of some neuromuscular diseases. Fasciculations of muscle, which are often best seen in the tongue, are a sign of denervation. Sensory abnormalities indicate neuropathy. Fatigable weakness is characteristic of neuromuscular junctional disorders. Myotonia is specific for a few myopathies.
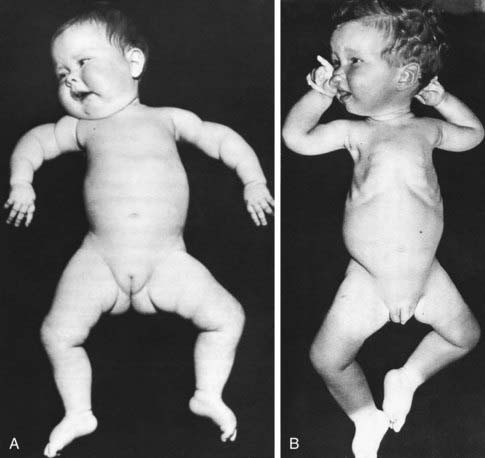
The thorax of infants with congenital neuromuscular disease often has a funnel shape, and the ribs are thin and radiolucent as a result of intercostal muscle weakness during intrauterine growth. This phenomenon is characteristically found in infantile spinal muscular atrophy but also occurs in myotubular myopathy, neonatal myotonic dystrophy, and other disorders (Fig. 599-1). Because of the small muscle mass, birth weight may be low for gestational age.
Generalized hypotonia and motor developmental delay are the most common presenting manifestations of neuromuscular disease in infants and young children (Table 599-2). These features can also be expressions of neurologic disease, endocrine and systemic metabolic diseases, and Down syndrome, or they may be nonspecific neuromuscular expressions of malnutrition or chronic systemic illness (Table 599-3). A prenatal history of decreased fetal movements and intrauterine growth retardation is often found in patients who are symptomatic at birth. Developmental disorders tend to be of slow onset and are progressive. Acute flaccid paralysis in older infants and children has a different differential diagnosis (Table 599-4).
Table 599-2 PATTERN OF WEAKNESS AND LOCALIZATION IN THE FLOPPY INFANT
| ANATOMIC REGION OF HYPOTONIA | CORRESPONDING DISORDERS |
|---|



