Underlying conditions
Metabolic: rickets, renal osteodystrophy
Genetic disorders: osteogenesis imperfecta, neurofibromatosis
Neuromuscular: cerebral palsy, Charcot-Marie-Tooth arthrogryposis
Skeletal dysplasias
Benign tumors: fibrous dysplasia, Ollier’s, multiple hereditary exostoses
Malignant tumors
Inflammation: rheumatoid arthridities, hemophilic arthropathy
Vascular anomalies
Congenital
Congenital femoral deficiency (proximal focal femoral deficiency)
Congenital fibular deficiency (fibular hemimelia)
Congenital tibial deficiency (tibial hemimelia)
Tibial dysplasia: congenital pseudarthrosis, posteromedial bowing
Hemihypertrophy
Amniotic band syndrome
Congenital knee dislocation
Developmental
Genu varum
Blount’s disease
Genu valgum
Acquired: sequelae and complications
Residual hip deformity: developmental dysplasia of the hip, slipped capital femoral epiphysis, Legg-Calve-Perthes, avascular necrosis
Posttraumatic: Cozen fracture
Post-infectious
Iatrogenic
Underlying Generalized Conditions
Metabolic Disorders
Certain metabolic disorders and endocrinopathies can affect the maturation of growth plate chondrocytes by the alteration of normal regulatory signals or by disturbance of the functional matrix components necessary for new bone formation. Some endocrine disorders such as hypothyroidism and growth hormone deficiency [1] and metabolic disorders including renal osteodystrophy [2] can mechanically weaken the physis, predisposing to pathologic conditions such as slipped capital femoral epiphysis (SCFE). These same disorders can render the chondrocytes more susceptible to compressive forces, leading to angular deformities.
Rickets
Rickets is a clinical manifestation of defective mineralization of the long bone physes due to inadequate levels of calcium or phosphate in children. Osteomalacia, the defective mineralization of osteoid at cortical and trabecular surfaces, is seen in both children and adults. There are many different forms of rickets that result in very similar radiographic and clinical manifestations (Table 1.2). Patients may have decreased longitudinal growth and angular deformities [3] (Fig. 1.1).
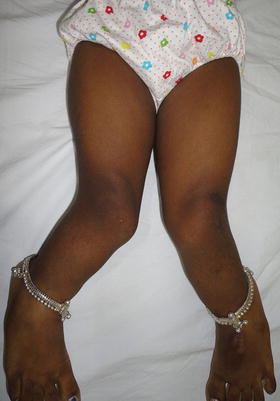
Table 1.2
Rickets and related disorders
Type | Cause |
|---|---|
Nutritional | Vitamin D deficiency |
Dietary calcium deficiency | |
Combination of both | |
Gastrointestinal | Poor absorption due to disease (gluten sensitivity, Crohn’s/ulcerative colitis, short gut) |
End-organ insensitivity | Insensitivity to vitamin D3 |
Vitamin D dependent | 1 alpha hydroxylase deficiency |
X-linked hypophosphatemia | PHEX gene defect, wasting of phosphate in kidney |
Renal tubule abnormality | Kidneys waste many molecules, particularly phosphate |
Hypophosphatasia | Alkaline phosphatase deficiency (gene mutation on chromosome 1) |
Renal osteodystrophy | Renal failure |
Fig. 1.1
Genu valgum in a 7-year old-female. The etiology was nutritional rickets. Image courtesy Dr. Hitesh Shah
Renal Osteodystrophy
Renal osteodystrophy is the combination of rickets and hyperparathyroidism that occurs secondary to end-stage renal disease. The findings of rickets are explained by the failing kidney being unable to produce a sufficient amount of 1, 25-dihydroxyvitamin D3. Additionally, the dysfunction of the tubular system decreases clearance of phosphate and causes secondary hyperparathyroidism, which manifests as brown tumors and subperiosteal erosions [3].
The deformity that results from these metabolic disorders depends on the stage along the physiologic varus/valgus spectrum that the child is at the age of onset of the disorder. The onset of nutritional rickets and inherited disorders is generally seen during infancy and therefore they produce genu varum. On the other hand, renal osteodystrophy usually causes genu valgum because acquired renal failure occurs later when children are more likely in the physiologic valgus age group [4].
Genetic Disorders
Osteogenesis Imperfecta
Osteogenesis imperfecta is caused by genetic mutations that affect the quantity or quality of collagen produced. Many distinct mutations have been identified and can thus create a wide spectrum of phenotypic manifestations. Those who are more severely affected have extensive bony fragility that leads to numerous fractures, short stature, and angular deformities of the long bones and the spine [5].
Neurofibromatosis
Neurofibromatoses (NF) are a group of genetic multisystem disorders involving products of all three germ lines: neuroectoderm, mesoderm, and endoderm. The most common and orthopedically relevant form is neurofibromatosis type 1 (NF1) previously known as von Recklinghausen disease, which involves predominantly peripheral nervous system although it involves cells of mesodermal origin as well.
NF1 is one of the most common autosomal dominant disorders with a prevalence rate of 1:4,000 [6]. It occurs almost equally in all ethnic groups. Approximately 50 % of all cases of NF1 are due to new mutations. The NF1 gene mutations have variable expression—the clinical manifestations of NF1 range from subclinical to severe. However, it has nearly 100 % penetrance in adult individuals. Thus, an adult carrying the mutation will eventually exhibit some clinical feature of the disease. The NF1 gene is located on the long arm of chromosome 17 (17q) and of relatively large size (350,000 base pairs). This may explain the large rate of new mutation associated with this disorder. The gene product neurofibromin acts as a tumor-suppressor gene [7].
Several distinguishing clinical features of NF1 help to make the diagnosis of the condition (Fig. 1.2; Table 1.3).
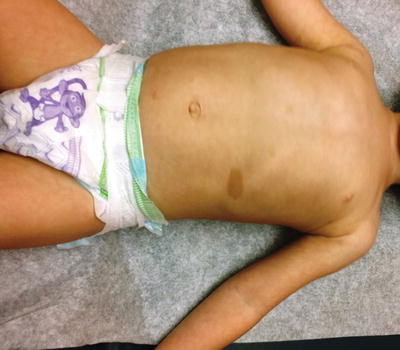
Fig. 1.2
A 3-year-old female patient with neurofibromatosis type 1 (NF1). Note the café au lait spots on the abdomen. Café au lait spots are the most easily noticeable feature of NF1
Table 1.3
Diagnostic criteria of neurofibromatosis type 1
More than six café au lait spots measuring at least 15 mm in adults and 5 mm in children |
Two or more neurofibromas of any type or one plexiform neurofibroma |
Freckling in the axillary or inguinal regions |
Optic glioma |
Two or more Lisch nodules (i.e., iris hamartomas) |
A distinctive bony lesion, such as sphenoid wing dysplasia, or thinning of the cortex of a long bone with or without pseudarthrosis |
A first-degree relative with NF-1, as suggested by these criteria |
There are a number of clinical issues associated with NF1 that are important from an orthopedic standpoint. These include tibial dysplasia and hemihypertrophy leading to leg length discrepancies and limb deformities, which are discussed later in the chapter.
Another important manifestation of orthopedic interest is development of spinal deformities. Spinal deformities are the most common orthopedic manifestation of NF1. True incidence of spinal deformities in NF1 is not known. It is quoted as from 2 to 36 % in the literature [8, 9]. In the NF clinic at our institution, it is 23 % [6]. Traditionally, two forms of spinal deformities are described in NF1: dystrophic and non-dystrophic. Non-dystrophic curves are similar to idiopathic curves with some exceptions. The dystrophic changes in NF1 include rib penciling, vertebral scalloping, vertebral wedging, spindling of transverse processes, and enlarged intervertebral foramina.
The spinal deformities tend to develop early in the life; therefore all preadolescent children with NF1 should be evaluated by scoliosis screening or the Adam’s forward-bend test to rule out the presence of a spinal deformity.
Neuromuscular Disorders
Cerebral Palsy
Depending on the distribution and severity of their underlying spasticity, children with cerebral palsy can have a wide array of rotational, angular deformities and joint contractures. Excessive femoral anteversion is quite common in spastic diplegia and is associated with increased tone in the medial hamstrings and adductors. Femoral anteversion can also be accompanied by compensatory tibial external rotation in diplegic or by internal tibial rotation in a hemiplegic patient. Muscle tone imbalances also cause deformities of the hips, knees, ankles, and feet [10].
Charcot-Marie-Tooth Disease
Charcot-Marie-Tooth disease (CMT) is the most common of the seven types of hereditary motor and sensory neuropathies (HMSN). The two general types are demyelinating and axonal, but they are not distinguishable by clinical exam. As research has identified specific locations of different genetic abnormalities, the number of described subtypes has increased. Type IA CMT disease is the most common subtype and occurs via autosomal dominant transmission of a peripheral myelin protein 22 gene duplication on chromosome 17. The X-linked dominant form of disease is second most common and is attributed to a mutation of the connexin-32 gene. In general, demyelination of peripheral nerves and degeneration of the dorsal root ganglia and dorsal column of the spinal cord in the central nervous system occur, leading to progressive muscle atrophy, loss of proprioception, and loss of deep tendon reflexes. The most common deformity is worsening cavovarus foot posture with claw toes (Fig. 1.3) due to selective intrinsic and extrinsic (tibialis anterior and peroneus brevis) muscle weakness and imbalance. The gastrocnemius-soleus complex is also weak and contracted, contributing to the cavovarus deformity and to the thin “stork leg.” The hands can be affected and, less commonly, patients can develop scoliosis or hip dysplasia [11].
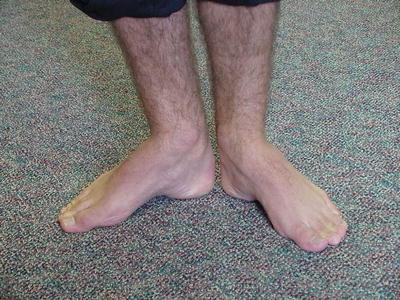
Fig. 1.3
A 17-year-old male patient with Charcot-Marie-Tooth disease. Note the cavus deformity of the foot bilaterally
Arthrogryposis Multiplex Congenita
Arthrogryposis multiplex congenita (multiple congenital contractures) is actually a symptom complex of deformed, rigid, and sometimes dislocated joints that can occur as part of at least 300 different disorders. Amyoplasia is the classic and most common form of arthrogryposis that involves all four extremities. It is a nonprogressive syndrome characterized by extremities that appear cylindrical, fusiform, or cone shaped due to atrophied or absent muscles, decreased subcutaneous tissue, and diminished skin creases. In the lower limb, the foot deformities commonly seen include a rigid clubfoot or vertical talus. The knees may have either flexion or extension contractures. The hips are also very often involved, usually with flexion, abduction contractures, and hip dislocation noted in 15–30 % of patients with amyoplasia. These clinical findings result from decreased fetal movement. Fetal akinesia is due to failure of skeletal muscle development, often related to abnormal development of anterior horn cells in the spinal cord [12].
Skeletal Dysplasias
Children with various skeletal dysplasias present clinically with short stature and variable growth disturbances in the trunk and extremities. Specific deformities are typical for each type of dysplasia and are caused by specific structural abnormalities within the bones and cartilage. The underlying genetic mutation and mode of transmission, autosomal dominant for most but not all, are known for the majority of bone dysplasias. In the lower extremities, specific angular deformities and involved joints are characteristic for each diagnosis.
Benign and Malignant Tumors
Tumors, both benign and malignant, have the potential to cause limb deformities. Solitary lesions, even benign simple bone cysts, can be complicated by growth arrest due to pathologic fracture, juxtaposition to the physis, or injury during therapeutic curettage or injection [13]. The risk of growth disturbance is inherently higher with malignant tumors as they are much more aggressive. Treatment of malignancies may require a wide resection, with or without an amputation, often including the nearby physis that leaves the child with an increasing length discrepancy with growth.
Polyostotic diseases such as fibrous dysplasia and Ollier’s disease are known to cause severe limb deformities. In fibrous dysplasia, expanding fibro-osseous tissue in the intramedullary canal causes repetitive microfractures, pain, and progressively bowed long bone deformities such as the classic shepherd’s crook appearance of the femur [14]. Ollier’s disease, a form of multiple enchondromatosis, produces islands of cartilage in the diaphyseal and metaphyseal regions that encroach upon the physis to cause inhibition of growth. Ollier’s is often unilateral, so it is not only associated with angular deformities such as distal femoral varus, but also tends to cause severe lower limb length discrepancies [15].
Inflammatory Disorders
Children with inflammatory arthritis such as rheumatoid conditions have chronic joint inflammation that causes pain and muscle spasms, which eventually results in hip and knee flexion contractures, growth disturbances, and angular deformities. However, these patients will have typical features such as small joint involvements in upper extremity during earlier stage of the disease, which provides a clue to the diagnosis (Fig. 1.4). In children with hemophilic arthropathy, the repeated intra-articular bleeding causes synovitis that ultimately results in joint contractures secondary to cartilage degradation from both direct compression from the hypertrophic synovial pannus and proteolytic enzyme release [16]. Along with the intra-articular hemorrhage, there is hyperemia that often stimulates growth, resulting in angular deformity if there is asymmetric involvement of the adjacent physis or limb length discrepancy if symmetric involvement occurs [17].
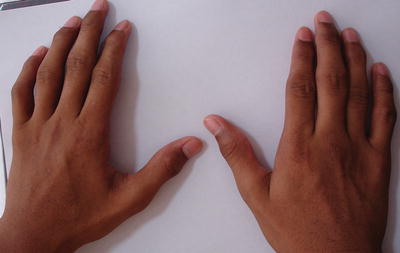
Fig. 1.4
A 14-year-old male patient with juvenile rheumatoid arthritis (JRA). Note enlarged small joints of the hands. Image courtesy Dr. Hitesh Shah
Vascular Malformations
Various generalized and local vascular malformations are another group of disorders that can cause significant limb length inequality of up to 10 cm in addition to hypertrophy in girth and skin alterations [18].
Congenital
Hemihypertrophy and Hemiatrophy
There are no clear guidelines in literature as to what constitutes hemihypertrophy or hemiatrophy. It is also difficult to diagnose these conditions as there exists a normal variation between two sides of the body. However, when clinically significant differences are found, it is important to investigate further, as most of them would follow under one of the conditions discussed below (Table 1.4).
Table 1.4
Common overgrowth and undergrowth syndromes
Common overgrowth syndromes | ||
Beckwith–Wiedemann syndrome | Macroglossia, macrosomia, midline abdominal wall defects, ear creases or ear pits, and neonatal hypoglycemia | Mutation of 11p15, leading to overactivity of the IGF-2 gene |
Proteus syndrome | Progressively worsening macrodactyly, hemihypertrophy, thickening of the skin, lipomas, subcutaneous tumors, verrucae, epidermal nevus and macrocephaly | Somatic activating mutation in AKT1 kinase in a mosaic manner |
Neurofibromatosis type 1 | Café-au-lait spots, cutaneous neurofibroma, axillary freckling, optic glioma, Lisch nodules, vertebral scalloping, tibial dysplasia | Mutation of NF1 gene located on chromosome 17 leading to disruption of Ras signaling controlled by neurofibromin |
Klippel-Trenaunay syndrome | Nevus flammeus (port-wine stain), venous and lymphatic malformations, and soft-tissue and bony hypertrophy of the affected limb | In some patients only: angiogenic factor VG5Q mutation. Translocation (5:11, 8:14) |
Common undergrowth syndromes
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||