Chapter 457 Enzymatic Defects
457.1 Pyruvate Kinase Deficiency
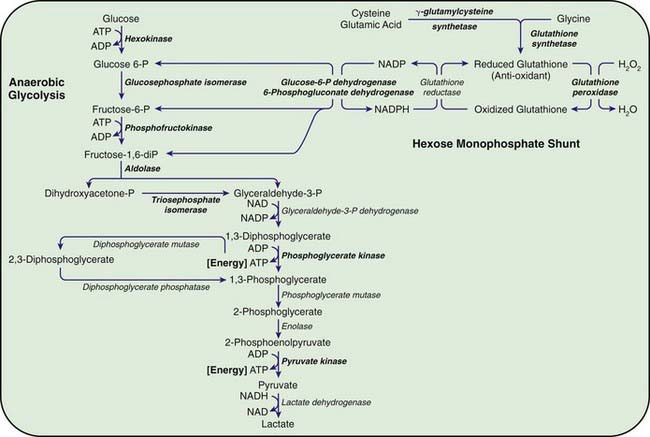
Congenital hemolytic anemia occurs in persons homozygous or compound heterozygous for autosomal recessive genes that cause either a marked reduction in red blood cell (RBC) pyruvate kinase (PK) or production of an abnormal enzyme with decreased activity. Generation of adenosine triphosphate (ATP) within RBCs is impaired, and low levels of ATP, pyruvate, and the oxidized form of nicotinamide adenine dinucleotide (NAD+) are found (Fig. 457-1). The concentration of 2,3-diphosphoglycerate is increased; this isomer is beneficial in facilitating oxygen release from hemoglobin but detrimental in inhibiting hexokinase and enzymes of the hexose monophosphate shunt. In addition, an unexplained decrease occurs in the sum of the adenine (ATP, adenosine diphosphate, and adenosine monophosphate) and pyridine (NAD+ and the reduced form of NAD) nucleotides, further impairing glycolysis. As a consequence of decreased ATP, RBCs cannot maintain their potassium and water content; the cells become rigid, and their life span is considerably reduced.
457.2 Other Glycolytic Enzyme Deficiencies
Chronic nonspherocytic hemolytic anemias of varying severity have been associated with deficiencies of other enzymes in the glycolytic pathway, including hexokinase, glucose phosphate isomerase, and aldolase, which are inherited as autosomal recessive disorders. Phosphofructokinase deficiency, which occurs primarily in Ashkenazi Jews in the USA, results in hemolysis associated with a myopathy classified as glycogen storage disease type VII (Chapter 81.1). Clinically, hemolytic anemia is complicated by muscle weakness, exercise intolerance, cramps, and possibly myoglobinuria. Enzyme assays for phosphofructokinase yield low values for RBCs and muscle.
Deficiencies of Enzymes of the Hexose Monophosphate Pathway
The most important function of the hexose monophosphate pathway is to maintain glutathione in its reduced state (GSH) as protection against the oxidation of RBCs (see Fig. 457-1). Approximately 10% of the glucose taken up by RBCs passes through this pathway to provide the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) necessary for the conversion of oxidized glutathione to GSH. Maintenance of GSH is essential for the physiologic inactivation of oxidant compounds, such as hydrogen peroxide, that are generated within RBCs. If glutathione, or any compound or enzyme necessary for maintaining it in the reduced state, is decreased, the SH groups of the RBC membrane are oxidized and the hemoglobin becomes denatured and may precipitate into RBC inclusions called Heinz bodies. Once Heinz bodies have formed, an acute hemolytic process results from damage to the RBC membrane by the precipitated hemoglobin, the oxidant agent, and the action of the spleen. The damaged RBCs then are rapidly removed from the circulation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


