9 Endocrinology
Normal Growth
Growth is influenced by many factors including overall health, heredity, gender, and environmental factors such as nutrition. Growth is rapid during the first year of life and then slows between 1 and 2 years of age (Table 9-1). After 2 years of age, linear growth continues to decline slowly, averaging a growth rate of approximately 2.0 inches (5 cm) per year until it reaches a nadir just before the initiation of the pubertal growth spurt. This nadir has been referred to as the “prepubertal dip” or the “prepubertal deceleration.” The “pubertal growth spurt” occurs during puberty. There are noticeable differences in the growth pattern of girls and boys during puberty. Girls generally start puberty at a younger chronologic age than boys and their pubertal growth spurt happens at an earlier pubertal stage. The pubertal growth spurt is also shorter in duration and displays a lower peak growth velocity in girls compared with boys. This sexual dimorphism is responsible for the differences in adult mature height between males and females. The average mature height of males is 13 cm greater than that of females.
Table 9-1 Normal Growth Rates in Children
| Growth Rate (per Year) | ||
|---|---|---|
| Age | Inches | Centimeters |
| Birth to 1 yr | 7–10 | 18–25 |
| 1 to 2 yr | 4–5 | 10–13 |
| 2 yr to puberty | 2–2.5 | 5–6 |
| Pubertal growth spurt: girls | 2.5–4.5 | 6–11 |
| Pubertal growth spurt: boys | 3–5 | 7–13 |
Because stature varies among healthy children, incremental growth rate is one of the most important elements used to assess health in a child. Subnormal growth velocity can indicate endocrine and nonendocrine disorders. The most critical tool to evaluate normal and pathologic growth is the growth chart. In May of 2000, the Centers for Disease Control and Prevention (CDC) released updated growth charts based on a broad population sample combining many different growth studies (Fig. 9-1, A–D). These charts are readily available (http://www.cdc.gov/growthcharts). The charts not only define the 3rd and 97th percentiles for height and weight, but also characterize standards for head circumference and body mass index (BMI) defined as weight (kg)/height2 (m). In contrast to adults for whom specific BMI values are used to define overweight and obesity, age- and gender-specific BMI values are used to classify children as over- or underweight. Health care professionals can use established percentile cutoff points to identify underweight and overweight children (Table 9-2).
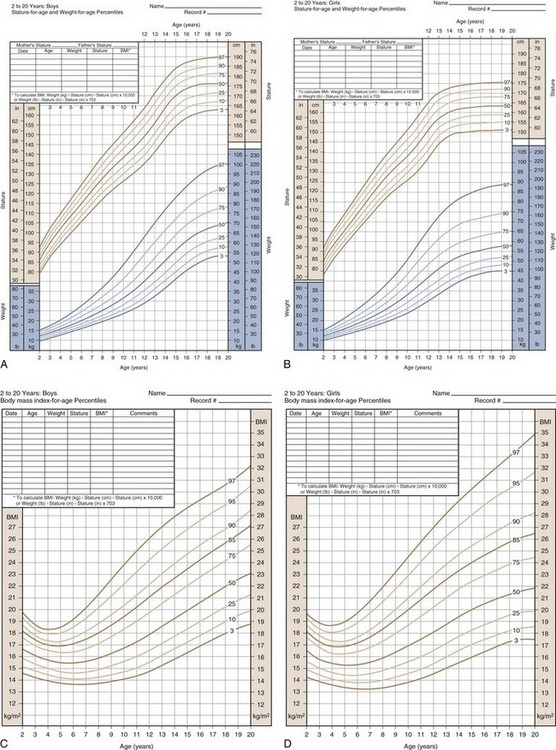
(Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion [2000]. Available online at http://www.cdc.gov/growthcharts.)
Table 9-2 Body Mass Index Cutoff Points
| Category | BMI for Age (Range) |
|---|---|
| Underweight | <5th percentile |
| Normal | 5th percentile to <85th percentile |
| At risk for overweight | 85th percentile to <95th percentile |
| Overweight | ≥95th percentile |
BMI, body mass index.
Short stature with normal body proportions and decreased growth velocity can be due to endocrine as well as nonendocrine disorders. Endocrine causes of short stature include growth hormone (GH) deficiency, GH resistance (GH receptor mutations or defects in insulin-like growth factor I [IGF-I] action), and hypothyroidism. Exogenous pharmacologic steroid therapy often leads to growth deceleration. Nonendocrine causes include chronic illness (e.g., renal tubular acidosis, celiac disease, or inflammatory bowel disease), genetic disorders, and undernutrition. One common cause of short stature in children is familial or genetic short stature. The genetic growth potential of a child is heavily influenced by the growth achieved by both parents and their relatives. The heritability of height has been estimated to be 0.7 to 0.8, rising to as much as 0.9 between identical twins. Common endocrine and nonendocrine causes of short stature are listed in Tables 9-3 and 9-4.
Table 9-3 Selected Nonendocrine Causes of Short Stature
ADHD, attention-deficit/hyperactivity disorder; CHARGE, coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness.
Table 9-4 Endocrine Causes of Impaired Growth and Short Stature
ADH, antidiuretic hormone; GH, growth hormone; IGF-I, insulin-like growth factor type I.
Children with constitutional delay typically have a period of decreased linear growth within the first 3 years of life. In this variation of normal growth and pubertal development, the rate of linear growth velocity and weight gain slow temporarily, often resulting in downward crossing of growth percentiles. By 2 or 3 years of age, linear growth resumes at a normal rate. Subsequently, children may grow either along the lower growth percentiles or beneath the curve but parallel to it for the remainder of their prepubertal years (Fig. 9-2). A family history of “late bloomers” or delayed puberty is common.
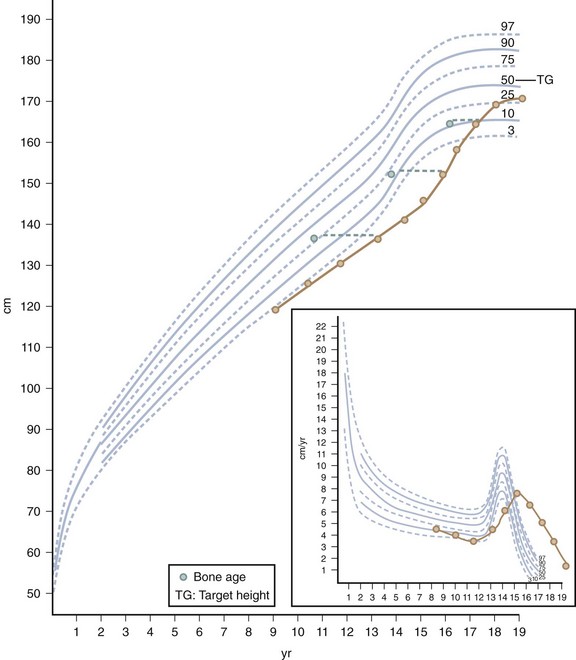
Pubertal Development
The pattern of timing of pubertal events for boys and girls is generally predictable (Fig. 9-3). For both boys and girls, mean ages for the onset of puberty vary among different ethnic groups and represent the combined influences of genetic and environmental factors. In boys, puberty, first evidenced by testicular enlargement, usually begins between 9 and 14 years of age. In girls, puberty, evidenced by breast development, usually begins between 8 and 12 years of age. Among white girls, the mean age of onset of breast development and pubic hair growth occurs at approximately 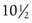 years of age, with menarche occurring at approximately
years of age, with menarche occurring at approximately 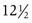 years of age. African-American females tend to enter puberty at an earlier age. For girls, increased BMI may be associated with premature adrenarche and earlier pubertal onset. Large-scale population studies have identified a secular trend for slightly earlier (by 2.5 to 4 months) ages at menarche, suggesting changes in the timing of the onset of puberty. Boys with rapid weight gain during childhood tend to have a later onset of puberty. The physiologic basis for this intriguing discordant effect of obesity on pubertal timing in boys versus girls is unexplained. These shifts, however, have generally not altered clinical practice guidelines regarding evaluation of “off-time” puberty.
years of age. African-American females tend to enter puberty at an earlier age. For girls, increased BMI may be associated with premature adrenarche and earlier pubertal onset. Large-scale population studies have identified a secular trend for slightly earlier (by 2.5 to 4 months) ages at menarche, suggesting changes in the timing of the onset of puberty. Boys with rapid weight gain during childhood tend to have a later onset of puberty. The physiologic basis for this intriguing discordant effect of obesity on pubertal timing in boys versus girls is unexplained. These shifts, however, have generally not altered clinical practice guidelines regarding evaluation of “off-time” puberty.
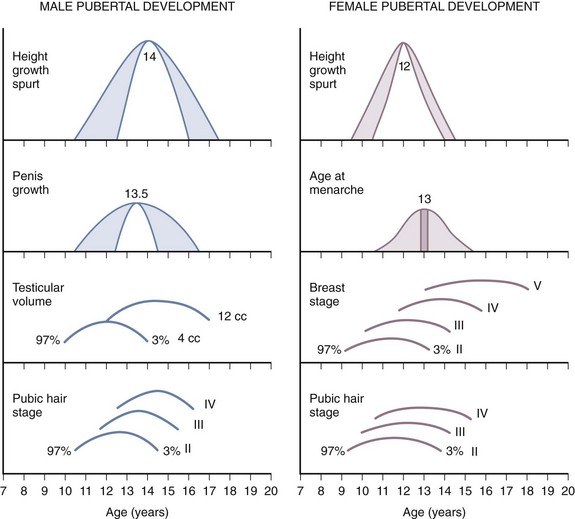
Figure 9-3 Schematic representation of the onset of male and female puberty.
(Modified from Johnson TR, Moore WM, Jeffries JE: Children are different: development physiology, ed 2, Columbus, Ohio, 1978, Ross Laboratories, Division of Abbott Laboratories, pp. 26-29. Used with permission of Ross Products Division, Abbott Laboratories, Inc., Columbus, Ohio 43215.)
Tanner Staging
To describe the onset and progression of pubertal changes (Fig. 9-4), boys and girls are rated on five-point scales. Boys are rated for both genital development and pubic hair growth, and girls are rated for breast development and pubic hair growth. In general, for healthy children, Tanner stages of puberty for genital development and pubic hair growth are congruent. Differences in pubertal staging may be useful to formulate the differential diagnosis for children with “off-time” puberty. Attention should be paid to the Tanner staging of genital and pubic hair development especially for the child with “off-time” puberty. Inconsistent staging assists with the initial differential diagnosis.
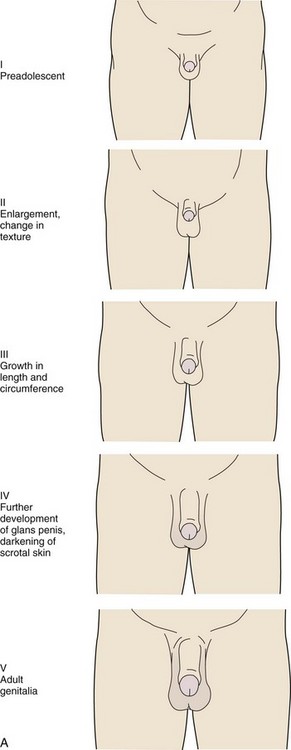
The stages for male genital development are as follows (Fig. 9-4, A):
Stage I (Preadolescent)—The testes, scrotal sac, and penis have a size and proportion similar to those seen in early childhood
Stage II—There is enlargement of the scrotum and testes and a change in the texture of the scrotal skin. The scrotal skin may also be reddened.
Stage III—Further growth of the penis has occurred, initially in length with some increase in circumference. There is also increased growth of the testes and scrotum.
Stage IV—The penis is significantly enlarged in length and circumference, with further development of the glans penis. The testes and scrotum continue to enlarge, and there is distinct darkening of the scrotal skin.
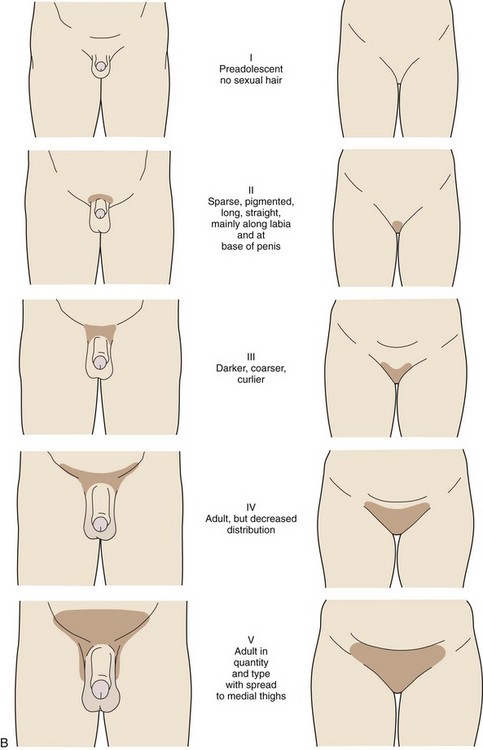
The stages in male pubic hair development are as follows (Fig. 9-4, B):
Stage I (Preadolescent)—Vellus hair appears over the pubes with a degree of development similar to that over the abdominal wall. There is no androgen-sensitive pubic hair.
Stage II—There is sparse development of long pigmented downy hair, which is only slightly curled or straight. The hair is seen chiefly at the base of the penis.
Stage III—The pubic hair is considerably darker, coarser, and curlier. The distribution of hair has now spread over the junction of the pubes.
Stage IV—The hair distribution is now adult in type but still is considerably less than that seen in adults. There is no spread to the medial surface of the thighs.
Stage V—Hair distribution is adult in quantity and type and is described as an inverse triangle. There can be spread to the medial surface of the thighs.
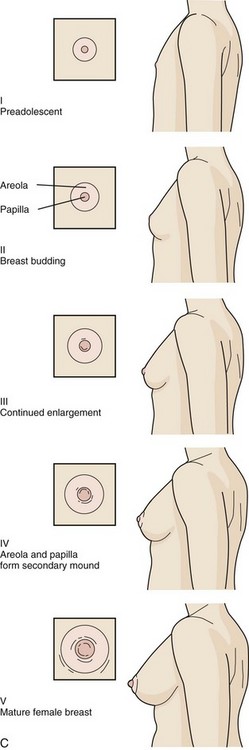
The stages in female breast development are as follows (Fig. 9-4, C):
Stage I (Preadolescent)—Only the papilla is elevated above the level of the chest wall.
Stage II (Breast budding)—Elevation of the breasts and papillae may occur as small mounds along with some increased diameter of the areolae.
Stage III—The breasts and areolae continue to enlarge, although they show no separation of contour.
Stage IV—The areolae and papillae elevate above the level of the breasts and form secondary mounds with further development of the overall breast tissue.
Stage V—Mature female breasts have developed. The papillae may extend slightly above the contour of the breast as the result of recession of the areolae.
Pubic hair growth in females is staged as follows (Fig. 9-4, B):
Stage I (Preadolescent)—Vellus hair develops over the pubes. There is no sexual hair.
Stage II—Sparse, long, pigmented, downy hair, which is straight or only slightly curled, appears mainly along the labia.
Stage III—Considerably darker, coarser, and curlier sexual hair appears. The hair has now spread sparsely over the junction of the pubes.
Stage IV—The hair distribution is adult in type but decreased in total quantity. There is no spread to the medial surface of the thighs.
Stage V—Hair is adult in quantity and type and appears in an inverse triangle of the classically feminine type. There is spread to the medial surface of the thighs, but not above the base of the inverse triangle.
The Hypothalamus and the Pituitary Gland
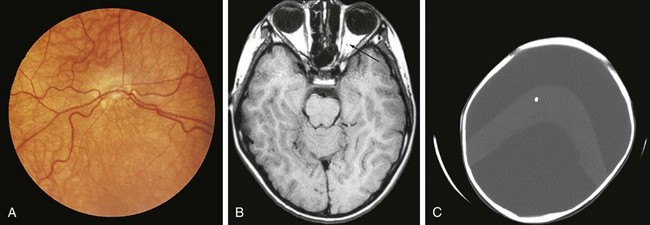
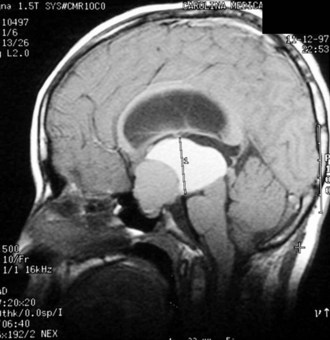
Both congenital and acquired abnormalities of pituitary function occur. Structural abnormalities of the central nervous system (CNS), such as septo-optic dysplasia (Fig. 9-5, A and B) and holoprosencephaly (Fig. 9-5, C), can interfere with pituitary function. Craniopharyngiomas and CNS tumors can be associated with acquired hypopituitarism (Fig. 9-6). Additional causes of acquired hypopituitarism include radiation therapy to treat CNS tumors, granulomatous infiltration, and traumatic interruption of the pituitary stalk.
Anterior Pituitary
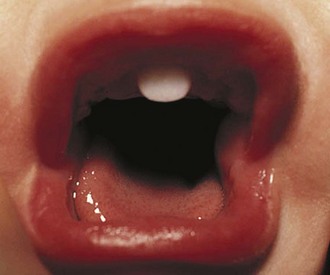
Children with midline defects have a higher incidence of hypopituitarism when compared with normal children. The child seen in Figure 9-7 has a single central incisor, an example of a midline abnormality associated with GH deficiency. Thus, specific alterations in physical appearance should alert physicians to a possible abnormality in anterior pituitary development potentially associated with secondary hormone deficiencies (e.g., thyroid-stimulating hormone [TSH] deficiency affecting thyroid function). The molecular etiologies for autosomal recessive, autosomal dominant, and X-linked disorders affecting anterior pituitary development and function have been elucidated.
Growth Hormone
GH influences linear growth and modulates several complex metabolic processes. GH secretion is regulated by the relative balance between the levels of GH-releasing hormone (GHRH) and somatostatin (Fig. 9-8). Both GHRH and somatostatin are secreted by the hypothalamus. The GH receptor is a single transmembrane protein. After the binding of GH to its receptor, a second GH receptor dimerizes with the first receptor to initiate the signal transduction process. GH generates direct effects via the GH receptor signal transduction pathway and secondary effects by promoting an increase in IGF-I and insulin-like growth factor-binding protein-3 (IGF-BP3).
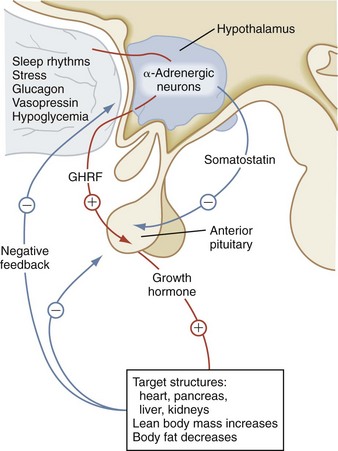
In children with hypopituitarism due to growth hormone deficiency, GH treatment markedly improves growth velocity. GH stimulates an increase in lean body mass, as well as a marked increase in the size of the heart, pancreas, liver, and kidneys. It has positive effects on carbohydrate, fat, and protein metabolism and causes a decrease in body fat. GH inhibits carbohydrate uptake by muscle. This diabetogenic effect of GH action is a known complication of GH hypersecretion. Typically, children with GH deficiency have normal birth weights and normal growth patterns during the first year of life, after which time their growth velocities decelerate. As seen in Figure 9-9, GH-deficient children have a characteristic “kewpie” doll appearance. They are often described as being “cherubic” because of their short stature, excess subcutaneous fat, retarded body proportion changes, and high-pitched voices. Infants with hypopituitarism may present in the early neonatal period with hypoglycemia or prolonged jaundice. On physical examination, male infants with hypopituitarism may have small penises due to concomitant luteinizing hormone (LH) deficiency.
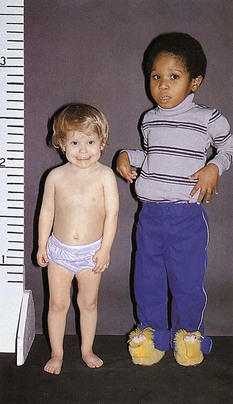
 old boy (right) is in the 50th percentile for height. The short 3-year-old girl (left) has GH deficiency.
old boy (right) is in the 50th percentile for height. The short 3-year-old girl (left) has GH deficiency.Adrenocorticotropic Hormone
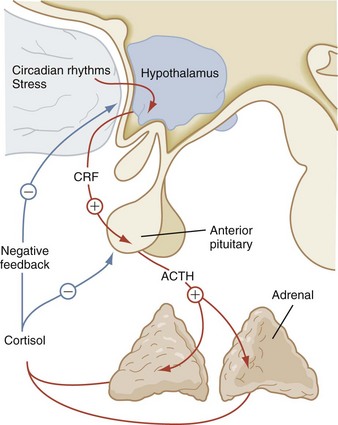
The corticotropin-related peptide hormones consist of adrenocorticotropic hormone (ACTH), α-melanocyte-stimulating hormone (α-MSH), and γ- and β-lipotropins (γ-LPH, β-LPH). These hormones are derived from a common precursor molecule, pro-opiomelanocortin. Within the subunit structure of β-LPH are the important neuroendocrine molecules α-, β-, and γ-endorphin and enkephalin. After posttranslational processing from this large precursor molecule, the secretion of ACTH is regulated by the level of corticotropin-releasing hormone (CRH), which is secreted by the hypothalamus. Cortisol secreted from the adrenal gland also influences ACTH secretion by negative feedback (Fig. 9-10). Prolonged pharmacologic glucocorticoid therapy suppresses the hypothalamic–pituitary–adrenal axis, with the potential for adrenal insufficiency as well as a significant impact on growth and development.
Gonadotropins
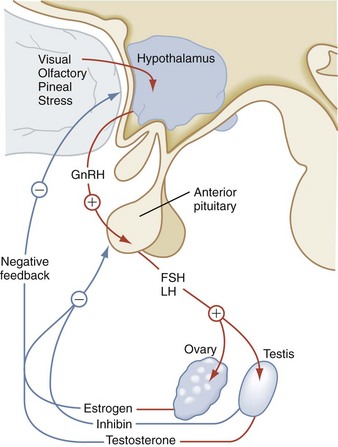
The primary actions of FSH and LH affect gonadal function. LH binds to the LH receptor on Leydig cells to stimulate testosterone synthesis and secretion. In the testes, FSH supports Sertoli cell development and spermatogenesis (Fig. 9-11). The ovary is characterized by a two-cell model for steroidogenesis. LH stimulates ovarian theca cells to synthesize androstenedione, which serves as the precursor for estradiol synthesis. Subsequently, in the ovarian granulosa cells, FSH increases expression of aromatase, the enzyme that converts androgens to estrogens. The negative feedback effect of sex steroids on LH and FSH production is dramatically emphasized in postmenopausal women and in individuals with gonadal failure, in whom marked elevations of these hormones occur. Inhibin B is produced by the gonads and inhibits FSH release.
Thyroid-stimulating Hormone
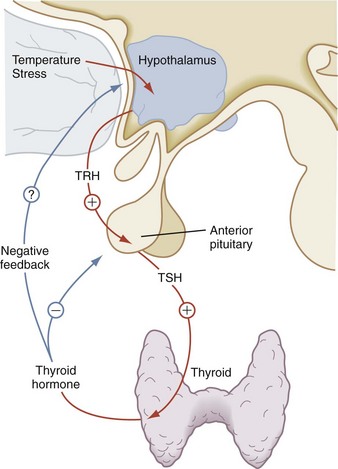
Thyroid-stimulating hormone (TSH) is the third glycoprotein hormone; its α subunit is identical to the α subunits of LH and FSH. Its specificity lies in its β subunit. TSH stimulates many aspects of thyroid function. Its major role is to promote the synthesis and secretion of thyroid hormone. Mediated by the TSH receptor, TSH increases the size of the thyroid cells, vascularity of the gland, iodide uptake, thyroglobulin synthesis, and thyroid hormone secretion. The rate of TSH secretion appears to be determined by the level of circulating thyroid hormone and by the hypothalamic hormone, thyrotropin-releasing hormone (TRH), as seen in Figure 9-12. Negative feedback of TSH secretion by circulating thyroid hormone occurs mainly at the pituitary level.
Posterior Pituitary
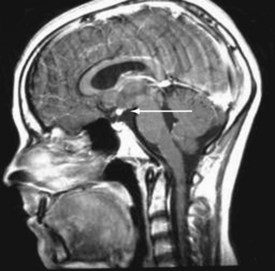
Vasopressin and oxytocin are two evolutionarily related peptides, each composed of nine amino acids. These hormones are synthesized in the hypothalamus and stored in the posterior pituitary gland. Expression of vasopressin and oxytocin genes occurs in the hypothalamic paraventricular and supraoptic nuclei. On magnetic resonance T1-weighted images, the posterior pituitary has a characteristic high signal intensity. The presence of this high signal intensity adjacent to the median eminence with absence of the normal pituitary bright spot within the sella on T1-weighted images is evidence of an ectopic posterior pituitary. An ectopic posterior pituitary is often associated with anterior pituitary hormone deficiencies, but typically these patients do not have diabetes insipidus (Fig. 9-13).
The Thyroid Gland
As seen in Figure 9-14, the thyroid gland usually is best palpated with the examiner behind the patient. After identification of the cricothyroid cartilage, the second and third fingers are moved laterally along the trachea just medial to the sternocleidomastoid muscles. Two distinct lobes are palpable; the right lobe is usually greater in size than the left lobe. When a goiter is present, these lobes may be quite easily identified (Fig. 9-15). The texture of the gland varies with hyperthyroidism and hypothyroidism, the former usually being soft and fleshy, and the latter usually firm or bosselated. Nodules can also be palpated and may be indicative of an adenoma or carcinoma. Because the thyroid is directly supported by the trachea, having the patient swallow will elevate and depress a palpable gland along with the trachea during the swallowing motion.
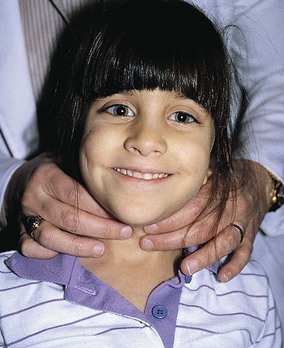
Figure 9-14 Examination of the thyroid gland. The thyroid gland is best palpated with the examiner behind the patient.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


