Diagnostic criteria are (1) based on laboratory measurements, symptoms of diabetes mellitus (DM) and random plasma glucose of ≥200 mg/dL, (2) fasting (≥8 hours), plasma glucose of ≥126 mg/dL, (3) a 2-hour plasma glucose of ≥200 mg/dL on an oral glucose tolerance test in the absence of acute illness, or (4) HbA1c > 6.5%.
Asymptomatic children should receive a provisional diagnosis of diabetes and have confirmatory testing with repeat testing on a different day.
Patients with fasting blood glucose of 100-125 mg/dL with symptoms of diabetes should have an oral glucose tolerance test (1.75 g/kg glucose, up to maximum of 75 g).
Autoimmune disease resulting from destruction of pancreatic beta cells
Characterized by absolute insulin deficiency
Classic clinical symptoms are polyuria, polydipsia, and weight loss.
Urgent referral of all patients with new-onset type 1 diabetes for initiation of insulin therapy and intensive education
Wearing medic alert bracelets by patients with this diagnosis is important.
Characterized by peripheral insulin resistance, impaired regulation of hepatic glucose production, and inadequate compensatory insulin secretory response, eventually leading to β-cell failure
Risk factors are obesity, family history, and PCOS.
Increased incidence in Native American, African American, Hispanic and Asian children of lower body weight
Screening should be done in children at high risk for type 2 diabetes with a fasting plasma glucose and HbA1c every 1-2 years beginning at age 10 or after onset of puberty.
There are different types of insulin preparations (Table 18-1)
Insulin is the first line treatment for all patients with T1DM and for those with T2DM with severe hyperglycemia or a HbA1c >8.5% or ketosis.
Suggested starting daily dosages for subcutaneous (SC) insulin at diagnosis, which are based on patient requirements:
<3 year = 0.3-0.4 U/kg/day
3-6 year = 0.5 U/kg/day
7-10 year = 0.6-0.8 U/kg/day
11-14 year = 0.8-1 U/kg/day
>14 year = 1-1.5 U/kg/day
Twice-daily injections: To be considered if poor compliance or if unable to count carbohydrates. Patients on this regimen should have fixed meal times and carbohydrate intake
2/3 total dose in AM: 1/3 lispro or aspart, 1/3 NPH
1/3 total dose in PM: ½ Humalog or NovoLog, ½ NPH
Basal bolus regimen—preferred insulin regimen in children. Can be given as multiple injections or via pump:
Allows for greater glycemic control and greater flexibility
Basal insulin dose is started as ½ total daily dose: once daily dose of glargine (Lantus) or twice daily dose of detimir (Levimir)
Remaining total daily dose is given with short-acting insulin (lispro or aspart) with meals—based on carbohydrate intake
Continuous subcutaneous insulin infusion (insulin pump): Give 80% of the basal insulin dose used on multiple SC injection over 24 hours. Mealtime boluses of short-acting insulin are given via the pump based on carbohydrate intake and premeal blood glucose values. With only short-acting insulin present in the pump, disruption of insulin delivery can be associated with ketosis and even diabetic ketoacidosis in a period of several hours; equivalent glycemic control can be obtained with basal bolus insulin and insulin pump with good compliance.
TABLE 18-1 Time Course of Action of Human Insulin Preparations | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
First line of treatment for adolescents with T2DM without ketosis.
It should be done before meals, bedtime, or if symptoms of low blood glucose occur. Middle-of-the-night (2 AM) glucose levels should be obtained at the onset of therapy with changes of PM or basal insulin doses.
Caloric requirements:
Up to age 10: 1,000 kcal + 100 kcal/year
After age 10: for females: 45 kcal/kg/day; for males: 55 kcal/kg/day
Tight dietary control is best achieved when patients count carbohydrates.
1 carbohydrate unit = 15 g of carbohydrate
TABLE 18-2 Hemoglobin A1c Values and Corresponding Blood Glucose Levels | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
If BG ≥ 300 or patient vomiting, he/she should check urine for ketones.
Hemoglobin (Hgb) A1c levels provide estimate of an average of blood glucose levels over the 3 months preceding measurement (Table 18-2). Monitor every 3-4 months. Target HbA1c in children is ≤7.5%, but may be individualized.
Microalbumin, eye exam, and monofilament exam annually in T1DM with ≥5 year duration or once in puberty and in all T2DM starting at diagnosis.
Lipids.
T1DM: every 5 years from 8-18 and annually after 18 years.
T2DM: annually after diagnosis.
Hypoglycemia is the most common complication of diabetes management and is the limiting factor of adequate glycemic control.
Symptoms are shakiness, sweatiness, nervousness, headache, irritability, confusion, and seizures.
Treat mild-to-moderate hypoglycemia with 15 g of fast-acting sugar, such as 4 oz juice or glucose tablets. Recheck blood glucose 15 minutes later.
Treat severe hypoglycemia (loss of consciousness or seizures) with glucagon 1 mg intramuscularly (if <20 kg, give 0.5 mg intramuscularly).
Hypoglycemia unawareness is the lack of hypoglycemic symptoms and adequate responses to hypoglycemia. This may develop in patients with tight diabetes control and recurrent hypoglycemia or after exercise. Will resolve with hypoglycemia avoidance.
Diabetic ketoacidosis (DKA) is characterized by serum glucose >200 mg/dL, ketonemia (>3 mmol/L) or ketonuria, dehydration, and serum pH < 7.3 or serum bicarbonate <15 mEq/L.
T1DM: new onset, insulin omission, illness
T2DM: severe illness, traumatic stress, or use of some antipsychotic agents
Patients with a range of symptoms that may be present with mild to severe DKA: vomiting, deep-sighing respirations (Kussmaul) with acetone odor, abdominal pain, and somnolence or loss of consciousness
Those with new-onset DM or ongoing poor glycemic control: also a history of polyuria, polydipsia, polyphagia, nocturia, and weight loss
Rapid assessment: blood glucose and urine ketones
Initial studies: basic metabolic profile (BMP), venous blood gas, complete blood count (CBC), Hgb A1c, urinalysis, electrocardiogram (ECG) if potassium is abnormal, blood and urine culture if temperature >38.5°C or signs of infection:
Anion gap (mEq): (Na – (Cl + HCO3)); normal: 8-12
Corrected Na: Na + [(glucose -100)/100] × 1.6
Plasma osmolarity: 2(Na) + Glucose/18 + blood urea nitrogen/2.8
Patients with DKA have plasma osmolarity >300 mOsm/L.
Characteristics: pH > 7.3, HCO3 >15 mmol/L, and moderate to large ketones
Often, outpatient treatment is appropriate.
Monitor BG and Ketones every 2 hours from insulin dose. Repeat dosing if ketones persist moderate or large.
Give additional short-acting insulin (lispro and aspart) every 2-3 hours:
Moderate urine ketones: usually 5%-10% of total daily dose
Large urine ketones: usually 10%-20% of total daily dose
If blood sugar is <150 mg/dL, it may be necessary to give additional sugary drinks to bring the blood sugar up before additional insulin.
Increase oral fluid intake to compensate for increased urinary losses and help clear ketones.
If patient is on an insulin pump and unable to clear ketones, give additional bolus of short-acting insulin by SC injection and change the pump site.
If concomitant hypoglycemia results from gastrointestinal (GI) disease, consider SC glucagon rescue therapy with 1 unit (10 µg)/year of age, starting at 2 units and up to 15 units (150 µg).
If patients are unable to clear ketones, or they have labored breathing, confusion, or lethargy, refer them to the emergency department for further care.
Characteristics include persistent emesis, high levels of ketones, pH 7.2-7.3, and HCO3 10-15 mmol/L.
Often, patients are managed in the emergency department or short-stay unit.
Intravenous (IV) hydration is often necessary.
Start 0.1 U/kg/hr regular I.V. insulin drip with hourly BG monitoring until anion gap closes. If unable to start drip consider giving 0.1 U/kg short-acting insulin every 2-3 hours, or 10%-20% of total daily dose, or regular insulin q2-4h or 2 times usual BG correction dose.
Admit if not resolving after 3-4 hours (i.e., Anion gap not closing and/or unable to take oral fluids), if newly diagnosed, or if the ability of caregivers is questionable.
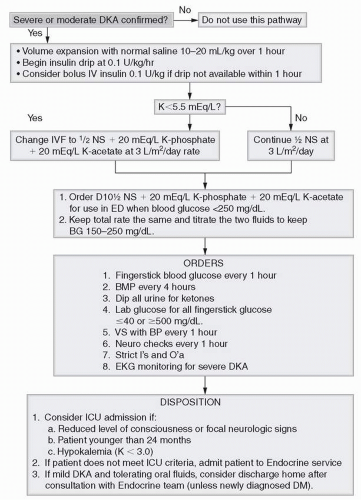 Figure 18-1 Algorithm showing the management of diabetic ketoacidosis (DKA). |
Characteristics include high levels of ketones, pH < 7.1, HCO3 < 10 mmol/L, pH < 7.2, or mild to moderate DKA along with other organ system impairment, such as altered mental status, impaired renal function, or respiratory distress.
Admit for therapy and intensive monitoring (fingerstick blood glucose q1h, BMP q4h, dipstick of all urine for ketones, vital signs with blood pressure q1h, neurology checks q1h, strict input/output).
Consider intensive care unit admission if patient has reduced level of consciousness or focal neurologic signs, age <24 months, or a potassium level <3.0 mg/dL.
TABLE 18-3 Summary of DKA Management | ||||||
|---|---|---|---|---|---|---|
| ||||||
Simple hydration frequently causes a 180-240 mg/dL drop in glucose.
Volume expansion (first phase [if poor perfusion or hypotension]): normal saline (NS) 10-20 mL/kg over 1 hour and then reassess volume status
Rehydration (second phase): ½NS plus potassium acetate plus potassium phosphate (see later discussion) at 3 L/m2/day:
Decrease to 2.5 L/m2/day if there are concerns about the risk of cerebral edema.
When blood glucose is <250 mg/dL, change to D5½NS. (Have D10½NS + potassium acetate + potassium phosphate available for use when blood glucose <250 mg/dL. Keep the total rate the same, and titrate the two fluids to keep blood glucose from 150 to 250 mg/dL.)
Once urine output is established and potassium is <5.5 mEq/L, start potassium administration.
Potassium level falls with correction of acidosis, decreased blood glucose, and initiation of insulin.
Add potassium 30-40 mEq/L to IV fluids as potassium chloride, potassium phosphate, and/or potassium acetate (i.e., ½NS + 20 mEq/L, potassium phosphate + 20 mEq/L potassium acetate at 3 L/m 2/day).
Volume expansion should be initiated before insulin administration.
Initiate the insulin drip at 0.1 U/kg/hr.
If the blood glucose is <150 mg/dL and the patient remains acidotic, do not stop the insulin drip, but increase the dextrose. If the acidosis is resolving (pH > 7.3, HCO3 > 15 mmol/L), the insulin infusion rate can be reduced to 0.08 or 0.05 U/kg/hr, especially if 10% dextrose is required to keep glucose above 150 mg/dL.
Change to SC insulin when patient is able to take oral fluids, the pH is >7.25, or HCO3 is >15 mmol/L, and the anion gap has closed. Consider administration of PM Lantus during treatment of DKA to provide basal insulin, which facilitates discontinuation of insulin drip at the appropriate time.
This is the most common cause of death during DKA in children (0.4%-1% of cases).
Anticipate cerebral edema in the first 24 hours after initiation of treatment. Always have mannitol available during the first 24 hours in patients with severe DKA.
Symptoms are change in affect, altered level of consciousness, irritability, headache, equally dilated pupils, delirium, incontinence, emesis, bradycardia, and papilledema.
Treatment
Cerebral edema is a medical emergency and immediate intervention is necessary.
Cerebral edema is a clinical diagnosis. Brain computed tomography (CT) is not indicated before treatment or to establish diagnosis, but consider CT to evaluate for thrombosis or infarction in addition to cerebral edema.
Mannitol 0.5-1 g/kg IV push over <30 minutes.
Decrease IV infusion rate to 2-2.5 L/m2/day.
Consider hyperventilation and dexamethasone.
There is a normal process during fasting to maintain fuel supply to the brain.
Normal fasting adaptation includes (1) hepatic glycogenolysis (when glycogen stores are depleted: >4 hour fast in infants and >8 hour fast in children), (2) hepatic gluconeogenesis, and (3) hepatic ketogenesis.
Hypoglycemia does not represent a single entity but is a defect in these major adaptive pathways.
Clinical hypoglycemia is defined as presence of Whipple’s Triad: (1) signs and symptoms of hypoglycemia, (2) documented low BG, and (3) resolution with carbohydrate intake.
A plasma glucose level below 50 mg/dL is recognized as the glycemic threshold for hypoglycemia.
Infants: cyanotic spells, apnea, respiratory distress, refusal to feed, subnormal temperature, floppy spells, myoclonic jerks, somnolence, and seizures
Children: tachycardia, anxiety, irritability, hunger, sweating, shakiness, stubbornness, sleepiness, and seizures
Infants and children often cannot recognize or communicate symptoms and recurrent hypoglycemia may blunt symptoms and hormonal responses.
A good history is crucial when evaluating hypoglycemia.
Information to know is the age of patient, gestational age and birth weight (for infants), length of the fasting period, triggering event (e.g., fructose ingestion), glucose infusion rate (GIR), perinatal history, and comorbidities (e.g., liver disease, midline defects, etc.), potential ingestion of glucose lowering agents.
An actual laboratory blood glucose measurement, not a glucometer result, to confirm true hypoglycemia is very important.
The critical sample to diagnose the underlying cause generally must be obtained during a hypoglycemic episode or during a formal fast. This sample is obtained when blood glucose levels fall below 50 mg/dL:
Samples for blood glucose, serum HCO3, insulin, C-peptide, β-hydroxybutyrate, lactate, free fatty acids, cortisol, growth hormone, and plasma NH3 are obtained.
Urine for ketones is also obtained immediately following the hypoglycemia.
In patients who are being worked up for hypoglycemia, also obtain blood for plasma total and free carnitine, urinary organic acid profile, and plasma acylcarnitine profile (always do so before a formal fast).
During a normal response to a blood glucose level below 50 mg/dL, the insulin level should be undetectable (<2 µU/mL), β-hydroxybutyrate increased (2-5 mM), lactate reduced (<1.5 mM), free fatty acids increased (1.5-2 mM), and counterregulatory hormones increased.
Infants of diabetic mothers
This manifests as transient hypoglycemia as a result of hyperinsulinemia following chronic exposure to elevated blood glucose in utero. Infants are usually macrosomic, and the hypoglycemia can last 3-7 days.
Treatment consists of frequent feeds or, if needed, supplemental IV glucose at a rate not to exceed 5-10 mg/kg/min.
Intrauterine growth retardation and perinatal stress
This can be manifested as hypoglycemia and usually persists for >5 days of life. Insulin levels may be inappropriately elevated.
Treatment involves frequent feedings, or most infants are responsive to diazoxide (5-15 mg/kg/day).
Infants taking β-blockers, which cause hypoketotic hypoglycemia because of suppression of lipolysis
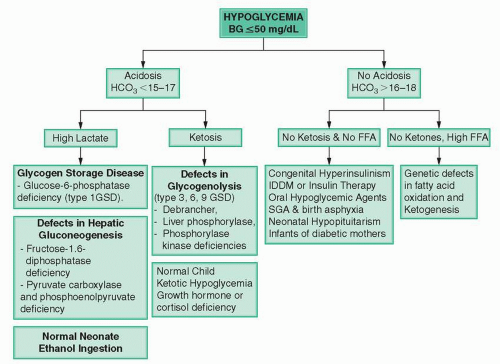 Figure 18-2 Algorithm showing the management of hypoglycemia. |
Hypoglycemia with lactic acidosis: inborn errors of metabolism
Glycogen storage disease type 1 (glucose-6-phosphatase deficiency)
Infants can develop hypoglycemia on day of life 1, although because of frequent feeds, this can go undiagnosed for months. Fasting tolerance is usually very short (2-4 hours).
Associated conditions include lactic acidemia, tachypnea, hepatomegaly, hyperuricemia, growth failure, hypertriglyceridemia, and neutropenia.
Treatment consists of frequent carbohydrate feeds, uncooked cornstarch (>1 year of age), limited fructose and galactose intake, and granulocyte-macrophage colony-stimulating factor.
Defects in hepatic gluconeogenesis (fructose-1,6-diphosphatase deficiency)
Patients usually develop hypoglycemia after fasting for 8-10 hours or after fructose ingestion.
Associated conditions include lactic acidemia and hepatomegaly.
Galactosemia (galactose-1-phosphate uridyl transferase deficiency)
This usually presents with jaundice without hepatomegaly and neonatal Escherichia coli-related sepsis.
Later on in life, patients can develop hepatomegaly, cataracts, developmental delay, ovarian failure, and Fanconi syndrome.
Treatment consists of galactose-restricted diet.
Hypoglycemia with lactic acidosis: alcohol intake or rubbing alcohol.
Normal newborns. Infants have poor ability to make ketones and gluconeogenesis in first 24 hours of life
Hypoglycemia with ketosis
Inborn errors of metabolism: glycogen storage disease types 3, 6, and 9 (debrancher, liver phosphorylase, or phosphorylase kinase deficiencies)
Fasting tolerance is usually 4-6 hours.
Patients can present with failure to thrive, hepatomegaly, cardiomyopathy, and myopathy.
Treatment consists of frequent feedings, low-free-sugar diet, and uncooked cornstarch.
Cortisol and growth hormone deficiency (hypopituitarism)
The incidence of hypoglycemia is ˜20%; beyond the neonatal period, this is usually associated with ketosis.
Fasting tolerance is usually 8-14 hours.
Treatment is adequate replacement therapy (8-12 mg/m2/day for hydrocortisone and 0.3 mg/kg/week for growth hormone).
Ketotic hypoglycemia
This occurs more commonly during the toddler and preschool age during periods of intercurrent illness, with poor oral intake or fasting periods of 10-12 hours. It is a diagnosis of exclusion.
Treatment involves frequent carbohydrate intake during periods of illness and avoidance of a prolonged overnight fast.
Hypoglycemia without acidosis (no ketosis; no elevated free fatty acids):
Congenital hyperinsulinism
The most common cause of persistent hypoglycemia of the newborn.
Time of onset, clinical features, fasting tolerance (0-6 hours), and therapy depend on the severity and type of disease or mutation. Patients usually do not present with failure to thrive.
Patients usually have high glucose requirements (10-30 mg/kg/min).
Patients respond to a glucagon stimulation (0.03 mg/kg to a maximum of 1 mg IV) with an increase in glucose >30 mg/dL within 15-30 minutes.
Different types include:
Recessive mutations of potassium channel genes (SUR 1, Kir6.2). Treatment is octreotide and subtotal pancreatectomy (98%); patients are unresponsive to diazoxide.
Dominant mutation of potassium channel genes. Treatment is subtotal pancreatectomy (98%).
Focal hyperinsulinism: focal loss of heterozygosity for maternal 11p and expression of paternally transmitted potassium channel mutations of either SUR 1 or Kir6.2. Treatment is focal resection; patients are unresponsive to diazoxide.
Dominant mutations of glutamate dehydrogenase: hyperinsulinism hyperammonemia syndrome. Treatment is diazoxide.
Dominant mutations of glucokinase. Treatment is diazoxide.
Recessive mutations of short-chain acyl-CoA dehydrogenase (SCHAD): abnormal metabolites in acylcarnitine profile and urine organic acids. Treatment is diazoxide.
Neonatal hypopituitarism. Clinical features associated with this condition are midline defects, microphallus, cholestatic liver dysfunction, and jaundice.
Furtive insulin or oral insulin secretagogue administration is characterized by hypoglycemia with high insulin levels but low C-peptide. When this is suspected, social work should be involved in evaluation of the case.
Post-Nissen dumping syndrome occurs in some infants following surgery for reflux disease.
Treatment consists of frequent feedings, and inhibitors of gastric motility as well as acarbose may be useful.
Hypoglycemia without acidosis (no or abnormally low ketosis but high free fatty acids)
Fatty acid oxidation and ketogenesis defects. Patients do not present in the neonatal period because fasting tolerance is 12-16 hours. The first episode is usually triggered by nonspecific illness.
The goal is to keep blood glucose above 70 mg/dL after a 7-hour fast and between meals.
Specific therapies include:
Dextrose: IV 0.2 g/kg bolus (2 mL/kg of 10% dextrose), followed by 10% dextrose continuous infusion (5 mL/kg/hr of 10% dextrose is approximately a GIR of 8 mg/kg/min in a newborn). Adjust rate to keep BG 70-150 mg/dL
Glucagon (only if insulin induced): 0.5 mg SQ or IV if <20 kg or 1 mg SQ or IV if >20 kg. Nausea and emesis are common side effects.
Diazoxide: 5-15 mg/kg/day divided into 2-3 doses. Start with maximum dose. Side effects: fluid retention and congestive heart failure
Octreotide: start at 2-10 µg/kg/day and may increase up to 50 µg/kg/day SQ divided q6-8h or continuous IV. Tachyphylaxis is a common problem, and this can cause suppression of other hormones such as glucagon, cortisol, growth hormone, and thyroid-stimulating hormone.
Uncooked cornstarch (glycogen storage disease type 1): 1-2 g/kg/dose in older infants.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


