Endocrine Disorders of the Newborn
Mary M. Lee
Thomas Moshang Jr.
From the moment of conception, physiologic endocrine processes are actively involved in growth and development of the human fetus. Disturbances in the interplay of these complex hormonal processes during intrauterine life can cause somatic or biochemical alterations in the fetus and newborn infant. Clinical disorders of endocrine function in the neonate therefore, can reflect an altered physiologic state in the fetus, the mother, or the fetal-maternal unit. Moreover, the occurrence of these perturbations of endocrine function at varying stages of fetal development results in diverse clinical manifestations. Knowledge of the ontogeny of the endocrine glands and their physiologic function during fetal development facilitates understanding disorders of endocrine function in the newborn.
DISORDERS OF SEXUAL DIFFERENTIATION
Normal Sexual Differentiation
The normal regulation of sexual differentiation is broadly illustrated in Fig. 39-1. All embryos are initially undifferentiated with a bipotential gonad and the anlagen for both male and female reproductive tracts and genitalia (1). Differentiation of the gonads as testes or ovaries dictates the subsequent development of the internal and external genitalia. The gonad forms when germ cells migrate from the dorsal endoderm of the yolk sac to populate the genital ridges. At the fifth to sixth week of gestation, these primitive bipotential gonads consist of both cortical (ovarian) and medullary (testicular) components. The genital ridge is composed of three cell types: (a) germ cells destined to become prespermatogonia in the male or oogonia in the female, (b) supporting epithelial cells destined to become Sertoli cells (male) or granulosa cells (female) and (c) mesenchymal cells destined to become the steroid-producing Leydig cells (male) or theca cells (female). Development of the supporting cells as Sertoli or follicular are a critical determinant in whether the germ cells differentiate as spermatogonia or oogonia.
Of the genes that are critical for gonadal development, mutations in two have been clearly associated with gonadal dysgenesis. Mutations of the Wilms tumor gene (2) (WT1), are associated with three related syndromes (the WAGR contiguous gene syndrome, and Denys-Drash and Frasier) that affect renal function and gonadal development and mutations in the transcription factor, steroidogenic factor 1 (3) (SF-1), cause agenesis of the adrenals and gonads.
Sexually dimorphic differentiation of the gonads and reproductive system commences when the testis-determining gene is first expressed. In 1959, Ford and colleagues determined that the Y chromosome was necessary for male development (4); in 1966, the critical region for testis determination was localized to the short arm of the Y chromosome (5); and in 1990, the primary testis-determining gene was definitively identified at Yp11.3 by positional cloning in patients with sex reversal (6). This gene, termed SRY (sex-determining region of the Y chromosome), is a member of the Sox family of transcription factors that all contain a high mobility group (HMG) DNA-binding motif (6). Activation of SRY initiates differentiation of the bipotential gonad as a testis. Loss of function mutations in SRY or a delay in its onset of expression can cause XY sex reversal with gonadal dysgenesis, although a gain of function mutation in SRY causes XX sex reversal associated with incomplete testicular development.
SOX9, a presumptive target for SRY, is a related HMG box gene that induces the supporting cells of the gonadal ridge to differentiate as Sertoli cells (7). Inactivating mutations of SOX9 cause the skeletal anomalies of camptomelic dysplasia that is associated with variable penetrance of 46, XY sex reversal and testicular dysgenesis (8). A role for additional downstream genes in testicular determination is supported by the absence of identifiable SRY or SOX9 mutations in
some cases of sex reversal and the association of sex reversal and dysgenetic gonads with other autosomal deletions.
some cases of sex reversal and the association of sex reversal and dysgenetic gonads with other autosomal deletions.
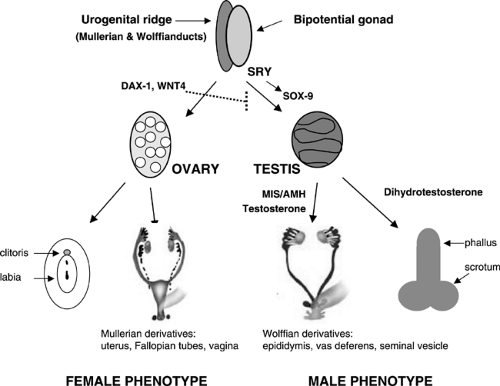 Figure 39-1 Schematic of the sex differentiation pathway. The urogenital ridge and gonad are initially undifferentiated. In male embryos, the induction of SRY expression initiates testis determination. The testicular hormones, MIS/AMH and androgens stimulate male phenotypic development of the internal and external genitalia. In female embryos, the absence of SRY in concert with the expression of genes that inhibit testicular determination enables the gonad to develop as an ovary. In the absence of androgens and MIS/AMH, the internal and external genitalia differentiate as female. |
At least two genes, Wnt-4, a locally secreted signaling glycoprotein, and Dax-1, a nuclear hormone receptor in the DSS region of the X-chromosome, are critical for ovarian development, perhaps by suppressing testicular genes. A duplication of either of these genes interferes with normal testicular development to cause a dosage sensitive form of 46, XY sex reversal (3,9).
Sexually dimorphic differentiation of the wolffian (male anlagen) and mullerian (female anlagen) internal genital tracts depends on the hormonal milieu established by the somatic cells. If SRY is expressed, the primary sex cords develop into testes and the somatic cells differentiate as Sertoli and Leydig cells. The Sertoli cells secrete Mullerian inhibiting substance (MIS), a 140-kd glycoprotein in the TGF-β family of proteins. MIS causes degeneration of the mullerian ducts by inducing apoptotic cell death of the ductal epithelial cells.
The Leydig cells secrete testosterone, which stimulates the wolffian ducts to differentiate into the vas deferens, seminal vesicle, and epididymis and virilizes the external genitalia.
Differentiation of the external genitalia requires the activation of testosterone by 5α-reductase-2 to its more active metabolite, dihydrotestosterone (DHT). DHT stimulates fusion of the urethral folds and the labioscrotal swellings to form the corpus spongiosa and scrotum. DHT also stimulates growth of the genital tubercle and prostate. Sexually dimorphic differentiation of the internal ducts and the external genitalia is complete by 12 weeks of gestation. Subsequently, during the latter part of gestation, the testes descend into the scrotum and the phallus enlarges as testosterone production increases under the stimulus of pituitary gonadotropins.
In 46, XX embryos, the primary sex cords become follicles by 10 weeks gestation and the germ cells differentiate to oogonia. Both X chromosomes are needed for maintenance of the ovary. In the absence of one X chromosome,
as in Turner syndrome, the ovaries form initially, but regress before birth. In females, fetal ovaries do not secrete MIS, thus the mullerian ducts differentiate to form the uterus, fallopian tubes and upper vagina. The absence of testosterone and DHT production by the fetal ovary cause the wolffian ducts to degenerate and enables the external genitalia to differentiate as female.
as in Turner syndrome, the ovaries form initially, but regress before birth. In females, fetal ovaries do not secrete MIS, thus the mullerian ducts differentiate to form the uterus, fallopian tubes and upper vagina. The absence of testosterone and DHT production by the fetal ovary cause the wolffian ducts to degenerate and enables the external genitalia to differentiate as female.
DISORDERS OF CHROMOSOMAL SEX
A number of sex chromosome aberrations have been reported (see Chapter 38): some are embryonic lethal (e.g., 45, YO), others cause minimal somatic or hormonal manifestations in the newborn (e.g., 47, XXY), and a few perturb gonadal and genital development (45, X/46, XY, 45, X0). In contrast to the autosomes, extra genetic material from the X chromosome can be tolerated with minor untoward effects as a result of inactivation of the second and additional X chromosomes. Although ovarian formation and function are intact in patients with X polyploidy, early menopause may occur (10). In contrast, a Y chromosome is generally necessary for testicular development, although rare cases of sex-reversed 46, XX males with normal testicular function and male genitalia are reported (11).
The classic sex chromosomal anomalies occur relatively frequently as determined by newborn screening. In the New Haven Study, 47, XXY occurred once in 545 males, 47, XYY occurred once in 728 males, 47, XXX occurred once in 727 females, and 45, XO occurred once in 2181 female newborns (12). The incidence of 45, XO if higher than this, but leads to increased fetal demise and is found in 1 in 15 spontaneous abortions (13). The diagnosis of Turner syndrome, however, is made with greater frequency than the other sex chromosomal aberrations because of the associated somatic abnormalities.
Turner Syndrome
The classic and most common chromosomal abnormality is total loss of one X chromosome. Over 50% of girls with Turner syndrome have a 45, XO karyotype, 17% have mosaicism with an isochromosome 46, X, i(Xq), 8% are chimeras with 45, X0/46, XX, and the remainder have other forms of mosaicism with loss of X material (14). The presence of a mosaic 46, XX cell line has little bearing on stature or somatic abnormalities, but does influence gonadal development. Goldberg and associates reported spontaneous female sexual development in 3 of 25 patients with mosaic karyotypes, but in none in those with 45, X0 (15).
The Turner phenotype in the newborn is secondary to lymphangiectasia and lymphedema. The webbed neck is most often seen as redundant folds about the posterior neck. The lymphedema involves the dorsa of the hands and feet. A host of associated somatic defects have been described in this syndrome (14,16), most of which become more evident with increasing age. The most common are triangular facies with low-set ears, high-arched palate, low hairline, shield-like chest with widespread and hypoplastic areolae, and cubitus valgus. Coarctation of the aorta is the typical cardiovascular abnormality; however the more benign condition of bicuspid aortic valves occurs more frequently. Miscellaneous renal malformations are observed. Skin manifestations include hemangiomas, cutis laxa, pigmented nevi, dysplastic nails, and tendency to keloid formation. Skeletal abnormalities include “beaking” of the medial tibial condyle, drumstick-shaped distal phalanges, vertebral anomalies, and short metacarpals (17). Dermatoglyphic abnormalities include palmar simian creases, distal axial triradius, and an increased number of digital ulnar whorls. Declining growth can manifest in young children and is the most consistent characteristic in the older child.
After diagnosis, screening for associated disorders such as cardiac and renal defects is needed (16). Therapy is focused on the specific developmental anomaly, such as coarctation of the aorta, and on providing education about potential associated problems, such as recurrent otitis media, chronic lymphocytic thyroiditis, and idiopathic hypertension. The incidence of mental retardation is slightly increased with specific X chromosome rearrangements. In most children with Turner syndrome however, cognition is normal with good verbal skills and selected spatial deficits.
A major concern for girls with Turner syndrome is extreme short stature with a mean adult height of 148 cm. Recombinant growth hormone increases final height and is approved for treatment of short stature in Turner syndrome. The combination of early use of growth hormone (before 5 years of age) and low dose estrogen replacement at an appropriate age is thought to give the best outcome in terms of height and pyschosexual development (18). Estrogen/progesterone therapy at the appropriate age is indicated for the treatment of sexual infantilism.
Questions regarding fertility may arise even in the newborn period because primary gonadal failure occurs in more than 90% of individuals with Turner syndrome. Women with Turner syndrome, however, have successfully carried offspring to term using donor oocytes, at a rate similar to couples with other causes of infertility (19).
The presence of Y material in the karyotype raises concerns about testicular elements that are at risk for malignant transformation. In patients with mosaicism that includes Y material, gonadectomy is therefore recommended, both to eliminate the risk for gonadoblastoma and to avoid the virilizing effects of hormonally active residual testicular elements (20).
DISORDERS OF GONADAL DETERMINATION
Disorders of gonadal determination can occur in association with autosomal or sex chromosomal anomalies and/or loss of function mutations or deletions of SRY and
SOX-97. Mutations have also been identified in other genes essential for gonadal formation such as WT-1 and SF-1. The clinical phenotype of these single gene mutations varies from complete gonadal dysgenesis to lesser degrees of testicular damage. Teratogens such as radiation, viruses, and drugs have also been implicated in in utero gonadal damage. Differentiation and development of the internal ducts and external genitalia in these infants depend on the timing and extent of insult to the developing gonad.
SOX-97. Mutations have also been identified in other genes essential for gonadal formation such as WT-1 and SF-1. The clinical phenotype of these single gene mutations varies from complete gonadal dysgenesis to lesser degrees of testicular damage. Teratogens such as radiation, viruses, and drugs have also been implicated in in utero gonadal damage. Differentiation and development of the internal ducts and external genitalia in these infants depend on the timing and extent of insult to the developing gonad.
Complete Gonadal Dysgenesis
Complete dysgenesis of the genital ridges results in normal female genitalia with no associated somatic findings, thus the diagnosis is often not suspected during infancy. Infants with 46, XY complete gonadal dysgenesis may be identified as a result of genitalia that are discordant with the prenatal karyotype. Affected girls tend to be tall with eunuchoid proportions and often present with primary amenorrhea and sexual infantilism. An autosomal recessive form of 46, XX gonadal dysgenesis is associated with sensory neural deafness (21). The majority of 46, XY gonadal dysgenesis is sporadic but familial forms can be sex-limited autosomal recessive, X-linked, or autosomal dominant in inheritance (22).
Partial Gonadal Dysgenesis
Incomplete loss of function of genes essential for testicular differentiation, or exposure to teratogens that damage the developing testis cause partial gonadal dysgenesis. Testicular loss after 9-10 weeks of gestation will not interfere with involution of the mullerian structures because the critical time for exposure to MIS is at 7 to 10 weeks, but will perturb mid-line fusion and development of the external genitalia, which is dependent on on-going testosterone production by the testes. Thus the external genitalia are female or severely undervirilized, but the gonads, uterus and fallopian tubes are absent and the wolffian structures are incompletely developed.
True Hermaphroditism
In true hermaphroditism, both ovarian and testicular elements are present. Findings may consist of an ovary on one side and a testis on the contralateral side, an ovary or a testis and a contralateral ovotestis, or two ovotestes (23). Most patients with true hermaphroditism have ambiguous external genitalia, although differentiation of the internal duct structures and external genitalia depend on the amount of functioning testicular tissue. In those reared as female, the testicular component of the gonad may secrete androgens at puberty to cause unwanted virilization, thus gonadectomy should be performed early. Although some patients have sex chromosome abnormalities, 46, XX is the most common karyotype, followed by 46, XY. The pathogenesis of true hermaphroditism is not well understood, but is not consistently linked to alterations in SRY expression.
DISORDERS OF PHENOTYPIC SEX
Disorders of phenotypic sex result when the anatomic development of the external genitalia does not correspond to the chromosomal and gonadal sex. The external genitalia may be truly ambiguous—that is, the sex of the infant cannot be ascertained by physical examination. Alternatively, the phenotype may be normal male or female, but inappropriate for the genotype. These conditions may be secondary to genetic defects affecting hormonal synthesis or action, problems in timing or regulation or hormonal secretion, defects in receptor binding or signaling defects, or teratogens or maternal hormones that perturb reproductive tract development. A genotypic (46, XY) male with testes and inadequate virilization falls in the category of male pseudohermaphroditism. A virilized genotypic (46, XX) female with ovaries is considered to have female pseudohermaphroditism.
Female Pseudohermaphroditism
The female fetus can be virilized by fetal adrenal androgens or maternal androgens transferred across the placenta. Exposure to androgens prior to week 12 of gestation results in fusion of the urogenital sinus and genital folds. Exposure to androgens after week 12 of gestation or after birth causes milder manifestations of clitoral enlargement, labial hyperpigmentation, and posterior labial fusion.
Congenital Adrenal Hyperplasia
The major cause of virilization in a female is congenital adrenal hyperplasia (CAH) as a result of steroidogenic enzyme defects in cortisol biosynthesis. The more common inherited enzymatic deficiencies of adrenal biosynthesis (21-hydroxylase, 11-hydroxylase, and 3β-hydroxysteroid dehydrogenase defects) all virilize the female. The gonads are ovaries, thus internal genital duct development is female—the wolffian ducts regress and the mullerian structures are normally formed. The excess adrenal androgens, however, promote fusion of the labia and the urogenital sinus, and phallic enlargement. Rarely, the virilization is severe enough to cause complete external masculinization. The 21-hydroxylase and 3β-hydroxysteroid dehydrogenase forms of CAH are complicated by associated mineralocorticoid deficiency that may present as a salt-losing adrenal crisis. The enzymatic defects causing CAH are more fully discussed in the section on adrenal disorders.
Drug-Induced Female Pseudohermaphroditism
A number of female newborns have been virilized by progestational agents used to prevent spontaneous abortion or other medications with androgenic potency (24). When these drugs are used during the first trimester, the labioscrotal folds are fused with formation of a urogenital
sinus and clitoromegaly. When fetuses are exposed after the first trimester, they develop only clitoral enlargement, without labioscrotal fusion. In contrast to untreated infants with CAH, there is neither progressive virilization nor continued acceleration of growth and skeletal maturation after birth. No medical intervention is needed as androgens are not elevated but surgical correction might be warranted. These children will feminize normally at puberty and achieve normal fertility.
sinus and clitoromegaly. When fetuses are exposed after the first trimester, they develop only clitoral enlargement, without labioscrotal fusion. In contrast to untreated infants with CAH, there is neither progressive virilization nor continued acceleration of growth and skeletal maturation after birth. No medical intervention is needed as androgens are not elevated but surgical correction might be warranted. These children will feminize normally at puberty and achieve normal fertility.
Virilizing Disorders in the Mother
Virilization of a female fetus by an androgen-producing maternal tumor is rare. These tumors almost always are caused by an ovarian lesion-arrhenoblastomas, Krukenberg tumors, luteomas, lipoid or stromal cell tumors, although adrenal adenomas are also reported (25). The tumors cause clitoromegaly, acne, deepening of the voice, decreased lactation and hirsutism in the mothers and are associated with elevated serum androgens and elevated excretion of urinary 17-ketosteroids (26).
Aromatase Deficiency
Rare genetic defects in the fetal or placental aromatase gene impair aromatization of maternal and placental androgens to estrogens and cause in utero elevations of androgens (27). Both fetal and maternal virilization can occur.
Idiopathic Female Pseudohermaphroditism
Idiopathic virilization may be caused by nonhormonal factors as exposure to androgens cannot be documented. This can occur in isolation or in conjunction with congenital anomalies of the gastrointestinal (GI) and urinary tracts that include imperforate anus, renal agenesis, urinary tract obstructions, urethrovaginal fistulas, and/or defective mullerian duct formation.
Male Pseudohermaphroditism
Incomplete masculinization of the male fetus can be as a result of a myriad of causes that disrupt either androgen action or the response of target tissues to androgens during sexual differentiation. The differential diagnosis of male pseudohermaphroditism is extensive, including enzymatic defects of testosterone synthesis, unresponsiveness to testos- terone action (androgen-resistance syndromes), hypothalamic or pituitary dysfunction, and vascular or teratogenic insult to the testis.
Defects in Testosterone Biosynthesis
Male fetuses are undervirilized when in utero testosterone production is reduced as a result of a genetic mutation in one of the testosterone synthetic enzymes (28). The phenotype varies according to the specific gene affected. Defects in 17α-hydroxylase/lyase and 17β-hydroxysteroid dehydrogenase primarily affect testosterone production although defects in enzymes needed for both testosterone and cortisol synthesis cause a form of CAH associated with incomplete masculinization. Steroidogenic acute regulatory protein (StAR) and 3β-hydroxysteroid dehydrogenase mediate early steps in cortisol and aldosterone synthesis. Mutations in either of these genes can result in a severe salt-losing syndrome and insufficient masculinization of the genitalia. The full details of these disorders, including diagnosis and treatment, are outlined in the section on adrenal hyperplasia.
Syndromes of Androgen Resistance
This is a group of disorders characterized by undervirilized genitalia with normal mullerian duct regression and normal testosterone synthesis (29). The term androgen resistance encompasses androgen receptor or postreceptor defects (androgen insensitivity syndrome [AIS]) and 5α-reductase deficiency in which the conversion of testosterone to its more active metabolite, DHT is affected. In both conditions, MIS is produced normally by the fetal testis and causes involution of the mullerian structures. In AIS, although testosterone is produced, the defect resides in the receptor or its signaling, thus target tissue response is compromised. Consequently, all aspects of male development mediated by androgens, including development of the wolffian structures and external genitalia are affected.
In contrast, in 5α-reductase deficiency, sufficient testosterone is produced for differentiation of the wolffian structures, but not for complete virilization of the external genitalia, which requires DHT (30). DHT is needed for mid-line fusion and phallic growth, thus patients with 5α-reductase deficiency typically have a blind vaginal pouch, a small phallic structure with chordee, a hooded prepuce, and perineoscrotal hypospadias. At puberty, the increase in secretion of testosterone and induction in expression of 5α-reductase and the androgen receptor in genital tissues stimulate growth of pubic hair, penile enlargement, and descent of the testes. 5α-reductase deficiency is suspected in 46, XY patients with perineoscrotal hypospadias and an elevated testosterone to DHT ratio of more than 35 under basal conditions and more than 74 after human chorionic gonadotropin (hCG) stimulation. The diagnosis is confirmed by finding reduced 5α-reductase activity in fibroblasts from genital skin.
Complete AIS (CAIS), an X-linked condition, was previously referred to as the testicular feminization syndrome (31). Affected patients have a 46, XY karyotype, normal female external genitalia with a blind vaginal pouch, absent wolffian and mullerian structures, and abdominal or inguinal testes. Unlike patients with 5α-reductase deficiency, virilization does not occur at puberty but peripheral conversion of the high testosterone concentrations to estradiol stimulates good breast development and estrogenization of the vaginal mucosa. Most patients have little pubic hair and some have total absence of sexual hair. In
all other respects, including height, habitus, voice, breast development, and gender identity, these individuals are feminine. The diagnosis is made in infancy or childhood as a result of female genitals that are discrepant with an 46, XY karyotype, or to the discovery of testicular tissue during hernia repair. Adolescent patients frequently present with primary amenorrhea. Genetic mutations of the androgen receptor are identified in only two thirds of individuals with suspected AIS. The gonads in CAIS have a 9% risk for malignant transformation to neoplasia, thus should be removed surgically.
all other respects, including height, habitus, voice, breast development, and gender identity, these individuals are feminine. The diagnosis is made in infancy or childhood as a result of female genitals that are discrepant with an 46, XY karyotype, or to the discovery of testicular tissue during hernia repair. Adolescent patients frequently present with primary amenorrhea. Genetic mutations of the androgen receptor are identified in only two thirds of individuals with suspected AIS. The gonads in CAIS have a 9% risk for malignant transformation to neoplasia, thus should be removed surgically.
A wide spectrum of phenotypes is observed in individuals with incomplete forms of androgen insensitivity. Partial or mild AIS (PAIS) can range from a female phenotype with clitoromegaly and posterior labial fusion to a male phenotype with infertility and sparse pubic hair. Sex assignment may be difficult in patients with PAIS. In some cases, assessment of responsiveness of the phallus to andro- gens is helpful.
OTHER CONDITIONS INVOLVING GENITOURINARY DEVELOPMENT
Male or Female Genital Phenotype Inconsistent with Genotype
A problem ensues at birth when the phenotype is inconsistent with the chromosomal sex determined by prenatal cytogenetic analysis. A repeat karyotype after birth may reveal an error in the prenatal cytogenetic analysis. A number of intersex conditions, however, can present at birth with external genitalia that are normal in appearance but discordant with chromosomal sex (Table 39-1). In general, these children should be raised according to the phenotypic sex.
Hypospadias and Cryptorchidism
Isolated hypospadias occurs in 0.8% of newborn infants and isolated cryptorchidism is present in approximately 5% of full-term and up to 15% of premature infants. Generally, neither condition by itself is associated with an endocrine abnormality. The incidence of intersex conditions however, is greater if the hypospadias is severe (on the shaft or perineum), or if the testes are nonpalpable. If cryptorchidism and hypospadias are both present, 25% of infants have a disorder of intersex.
TABLE 39-1 ETIOLOGY OF MALE OR FEMALE GENITAL PHENOTYPE INCONSISTENT WITH THE GENOTYPE | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Micropenis
Isolated micropenis with otherwise normally formed genitalia generally is not considered as ambiguous genitalia. This condition is associated with insufficient testosterone secretion during the third trimester. The evaluation of micropenis is discussed under hypopituitarism, the most common treatable cause of this condition.
EVALUATION
The evaluation of a newborn with ambiguous genitalia should be managed expediently by a team of experienced providers. Parents should be reassured that incomplete or excessive differentiation of the genitals occurred as part of a continuum in the developmental process and that the appropriate sex will be determined within several days. It is our general philosophy not to discuss pending studies in detail because there are occasions for gender assignment that are inconsistent with either chromosomal or gonadal sex and presentation of all data available enables a more cohesive explanation. As in any diagnostic problem, the approach to the child with ambiguous genitalia should begin with a thorough history, a careful physical examination, and appropriate laboratory and radiologic testing. Table 39-2 outlines the different causes of sexual ambiguity.
A history of drug ingestion, particularly in the first trimester, or recent androgenic changes in the mother might suggest the cause of female pseudohermaphroditism. First trimester infections or exposure to teratogens might suggest gonadal dysgenesis. A family history of an unexplained neonatal death or siblings with virilization or precocious puberty, might suggest the diagnosis of CAH although a history of female relatives with sexual infantilism suggests X-linked causes such as AIS.
A thorough physical examination is important, but on no account should a diagnosis be based on the physical
findings. The presence or absence of palpable gonads helps to differentiate the major categories of intersex conditions. In general, gonads lacking testicular elements will not descend below the inguinal region. Thus, a palpable gonad excludes the diagnosis of female pseudohermaphroditism in which the gonads are ovaries by definition. Measurement of the length and diameter of the penis is valuable both for prognostic information and also as a baseline if treatment is given to enlarge the penis. The urethral opening should be identified and the existence or absence of a vagina should be determined. The degree of fusion of the labial-scrotal folds and the presence of associated urinary or GI tract anomalies should be assessed.
findings. The presence or absence of palpable gonads helps to differentiate the major categories of intersex conditions. In general, gonads lacking testicular elements will not descend below the inguinal region. Thus, a palpable gonad excludes the diagnosis of female pseudohermaphroditism in which the gonads are ovaries by definition. Measurement of the length and diameter of the penis is valuable both for prognostic information and also as a baseline if treatment is given to enlarge the penis. The urethral opening should be identified and the existence or absence of a vagina should be determined. The degree of fusion of the labial-scrotal folds and the presence of associated urinary or GI tract anomalies should be assessed.
TABLE 39-2 ETIOLOGY OF AMBIGUOUS GENITALIA | |
|---|---|
|
The physical examination can help direct the laboratory and radiologic investigation. Certain tests are obtained as soon as it is apparent that there is sexual ambiguity, although others may be required at a later stage to make an accurate diagnosis (Table 39-3). For example, serum 17-hydroxyprogesterone and electrolytes are useful initial screening tests for congenital adrenal hyperplasia but other steroid precursors and genetic studies may help establish the specific diagnosis. In the newborn period, testosterone, follicle-stimulating hormone (FSH), and luteinizing hormone (LH) should be drawn to assess the hypothalamic-pituitary-gonadal axis. Serum testosterone can be elevated from either gonadal or adrenal production and should be interpreted in the context of the examination and other laboratory studies. Measurement of MIS may help determine the presence of testicular tissue (32). It should be stressed that sex assignment does not require that all studies leading to a final diagnosis be completed (e.g., the exact type of congenital adrenal hyperplasia may be important for genetic counseling and future prenatal diagnosis, but not for sex assignment). The karyotype can help determine whether the infant is a virilized female or an inadequately virilized male. This, however, should not be used as the primary criteria for sex assignment as other factors such as gonadal function, sensitivity to androgens, future sexual function and potential for fertility or pregnancy (even if by in vitro fertilization) are also critical.
Pelvic ultrasound to evaluate the internal genital structures and gonads should be performed by an experienced radiologist. Ultrasonography may identify nonpalpable gonads, and may be able to define the echogenic pattern as either ovarian or testicular tissue (33). The presence of a uterus indicates a lack of MIS action and probable absence of functioning testicular tissue in early gestation, and usually indicates that the child should be raised as a female. Conversely, the absence of mullerian structures implies the presence of functioning testicular tissue that secreted MIS at 7 to 9 weeks of gestation. This is consistent with sex-determining region Y (SRY) expression and suggestive of an XY karyotype, but is not a major determinant for male sex assignment. The karyotype, phallic size and degree of hypospadias, internal genital structures, gonadal pathology, and etiology of the intersex condition are all part of the equation in gender determination.
TABLE 39-3 STUDIES TO EVALUATE AMBIGUOUS GENITALIA | |
|---|---|
|
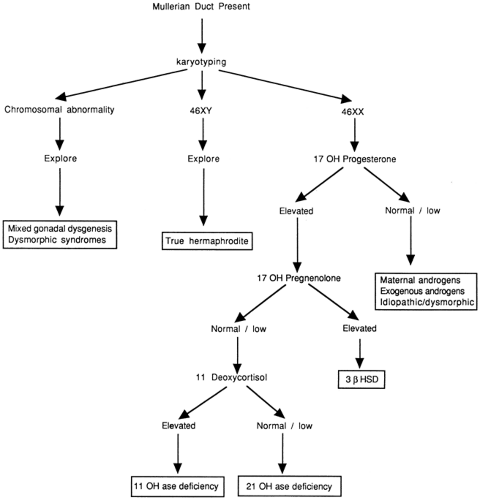 Figure 39-2 An algorithm for evaluating sexual ambiguity in infants with mullerian structures. |
To evaluate further the specific etiology of the sexual ambiguity, secondary studies may be necessary. The algorithms in Figs. 39-2 and 39-3, which are based on the initial ultrasound findings, delineate the steps that may be necessary to make a definitive diagnosis. These algorithms do not include patients with normal male or female phenotypes that are discordant with the genotype. Surgical exploration frequently will be required in cases of true hermaphroditism or partial gonadal dysgenesis but may be done at a later time. It should be stressed that the final histopathologic diagnosis is not necessary for sex assignment.
After the evaluation is complete, including consultations from an endocrinologist and an urologist, the appropriate sex assignment is determined by a consensus of opinion from the team that might also include a geneticist, a psychiatrist or psychologist, the pediatrician, and clergy or other support personnel. Parental input is also considered in the decision, especially in cases in which the appropriate sex assignment is uncertain. Gender identity, and future sexual function and fertility are major determining factors. The attending physician should discuss the condition fully with the parents, including expectations for future sexual function and fertility and whether any hormonal medications or surgery are recommended.
Gender assignment for most infants with ambiguous genitalia is straightforward when chromosomal sex and gonadal sex correlate with the internal structures. The external genitalia may require reconstructive surgery to improve function and cosmetic appearance. The timing of surgery has become controversial and highly politicized as a result of heightened concerns about ethical issues of informed consent by the child, the possibility of gender dysphoria and sex reassignment, and the risk for postsurgical loss of genital sensation. With little long-term data to support early vs. late reconstructive surgery, it may be prudent to postpone surgery until gender identity is clear and
to have the full participation of the family (and child) in the decision. Hormonal therapy may be required to induce secondary sexual maturation, but is usually not needed during the neonatal period. Rarely, as in cases of partial androgen resistance syndromes, true hermaphroditism, or mixed gonadal dysgenesis, gender assignment contrary to chromosomal or gonadal sex must be considered. In these cases, careful consideration must be given to the likelihood of gender role and sexual function as an adult (34).
to have the full participation of the family (and child) in the decision. Hormonal therapy may be required to induce secondary sexual maturation, but is usually not needed during the neonatal period. Rarely, as in cases of partial androgen resistance syndromes, true hermaphroditism, or mixed gonadal dysgenesis, gender assignment contrary to chromosomal or gonadal sex must be considered. In these cases, careful consideration must be given to the likelihood of gender role and sexual function as an adult (34).
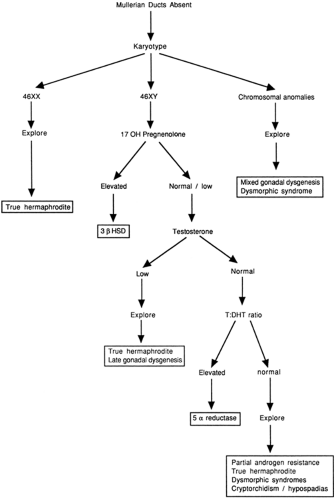 Figure 39-3 An algorithm for evaluating sexual ambiguity in infants without mullerian structures. |
Severe micropenis or agenesis previously warranted a female sex assignment despite the presence of testes or a normal 46, XY karyotype, but this approach is now under reappraisal. Recent reports of dissatisfaction with a female sex assignment in some 46, XY individuals with cloacal exstrophy or other nonhormonally mediated causes of aphallia reinforce the need to explore new paradigms for sex assignment. These paradigms will need to include other factors that affect adult gender identity such
as the role of prenatal hormones on central nervous system (CNS) sex differentiation.
as the role of prenatal hormones on central nervous system (CNS) sex differentiation.
DISORDERS OF THE HYPOTHALAMUS AND PITUITARY
Development of the Hypothalamic-Pituitary Axis
The hypothalamus arises by proliferation of neuroblasts in the intermediate zone of the diencephalic wall and formation of the supraoptic and periventricular nuclei. The anterior pituitary, or adenohypophysis, arises embryonically from an invagination of the oral ectodermal cavity called the Rathke pouch. This diverticulum arises at 3 weeks of gestation, and by 5 weeks has migrated upward to its final position and separated completely from the oral cavity. Concordantly, the posterior pituitary, or neurohypophysis, is formed from a downward invagination of the floor of the diencephalon. Neural fibers migrate from the hypothalamus down to the posterior pituitary to form the neurohypophyseal tract. The hypothalamus regulates the pituitary by secreting both stimulatory and inhibitory hormones. The stimulatory hormones include growth hormone-releasing hormone (GHRH), thyrotropin-releasing hormone (TRH), corticotropin-releasing hormone (CRH), and gonadotropin-releasing hormone (GnRH). The main inhibitory hormones are somatostatin, which inhibits growth hormone release and prolactin inhibitory factor, which inhibits prolactin release. In response to these hypothalamic hormones, the anterior pituitary secretes prolactin, growth hormone (GH), thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), LH, and FSH. The posterior pituitary secretes vasopressin and oxytocin. The hypothalamic and pituitary glands are functional after week 12 of gestation.
TABLE 39-4 ETIOLOGY OF DISORDERS OF THE HYPOTHALAMIC-PITUITARY AXIS | |
|---|---|
|
Most disorders of the hypothalamic-pituitary axis in the newborn period, except for the syndrome of inappropriate secretion of antidiuretic hormone (SIADH), are those of insufficiency related to malformations, trauma, infection or genetically inherited disorders, as outlined in Table 39-4. This differs from older children and adults who may have either functionally active tumors that secrete pituitary hormones or infiltrative disease or tumors that interfere with normal pituitary function.
DISORDERS OF THE ANTERIOR PITUITARY
Anterior pituitary dysfunction is difficult to detect in the newborn. The predominant features of anterior pituitary insufficiency are hypoglycemia, micropenis and, occasionally, cholestatic jaundice. The hypoglycemia may be quite severe and comparable to that seen in infants with congenital hyperinsulinism. The infants may even have a brisk glycemic response to glucagon causing further confusion (35). The cholestatic jaundice is initially unconjugated, then becomes predominantly conjugated, and will often only resolve after hormone replacement. There may be combined deficiency of multiple pituitary hormones or isolated deficiency of a single hormone. The molecular basis of multiple hormone deficiency is established for a number of genetic defects and was reviewed recently by Parks and associates (36). Evidence for the importance of the POU family of pituitary transcription factors in establishing pituitary lineages came from identifying the roles of Pit-1 in the Snell dwarf mouse (37), Prop-1 in the Ames dwarf mouse (38) and P-Lim knockout mice (39). In humans P-Lim is associated with deficiencies of all anterior pituitary hormones except ACTH in conjunction with retinal colobomas. Pit-1 mutations cause pituitary gland hypoplasia and deficiencies of GH, TSH, and prolactin. Prop-1 (Prophet of Pit-1) is essential for Pit-1 expression, and therefore has an identical clinical picture. Hypopituitarism in conjunction with optic nerve hypoplasia and absence of the septum pellucidum comprise the syndrome of Septo-optic dysplasia (SOD). Pituitary function can vary from intact to panhypopituitarism including diabetes insipidus. Mutations in a homeodomain protein, HESX-1, have been found in some patients with SOD associated with mild panhypopituitarism or isolated GH deficiency (40). SOD is often suggested by wandering nystagmus in the newborn, reflective of optic nerve hypoplasia and blindness.
Growth Hormone Deficiency
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


