Week
Day
Carnegie stage
Upper limb
Lower limb
4
24
11
Swelling appears in region of upper limb bud
28
13
Scattered blood vessels
Appearance of lower limb bud
5
32
14
Upper limb AER
Early marginal vessel
Early brachial plexus development
33
15
Hand plate appears
Lower limb AER
Humerus mesenchymal condensations
6
37
16
Humerus chondrification
Lumbosacral plexus
Radius and ulna mesenchymal condensations
Brachial plexus with radial, median, and ulnar nerves to the elbow
Early muscle masses
41
17
Finger rays (webbed)
Femur, tibia, fibula, and tarsus mesenchymal condensations
Radius, ulna, and metacarpal chondrification
7
44
18
Interdigital apoptosis
Femur, tibia, and fibula chondrification
Scapula and humeral head chondrification
Carpals and proximal phalanges chondrification
Trapezius innervated (accessory nerve)
Major muscles distinguishable
48
19
Middle phalanges chondrification
Shoulder and elbow interzones (joint cavity formation)
8
51
20
Distal phalanges chondrification
52
21
Humerus ossification
Radius ossification
All muscles distinguishable
Wrist and carpal interzones
54
22
Ulna ossification
Femur and tibia ossification
9
57
23
Scapula ossification
Fibula ossification
Intramembranous ossification of distal tip of distal phalanges
Tarsus and digits chondrification
The upper limb develops proximal to distal, starting from the trunk, and begins initially as a homogenous mass of undifferentiated mesenchymal cells. During limb outgrowth, musculoskeletal elements generally precede development of other elements such as nerves, vasculature, and lymphatics. Three distinct segments are identifiable in both the developing and mature limb: the stylopod (upper arm), zeugopod (forearm), and autopod (hand plate) (Fig. 1). Development of the three segments occurs both sequentially and concurrently. That is, the most proximal structures tend to be at a slightly more mature developmental stage than the more distal structures due to earlier onset of formation.
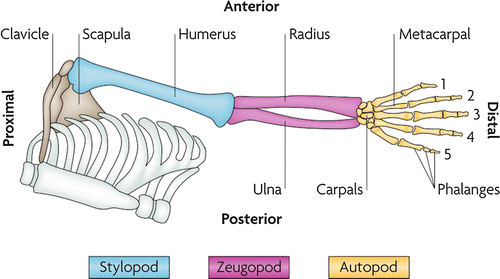
Fig. 1
Anatomy of the developing limb. The development of proximal structures precedes the development of more distal structures due to earlier onset of formation (Zeller et al. 2009)
A host of genes and molecular signaling work harmoniously together to ensure proper development of these structures. Much of these mechanisms and pathways remain to be worked out, but there is sufficient knowledge to be able to characterize defects in the context of basic embryological events that define the complex, coordinated process of limb development. Whereas early developmental work focused on the morphology of these embryological events, the emphasis is now placed on understanding the molecular basis of these changes, enabling detailed understanding of genotype-phenotype correlations that may have substantial clinical implications.
In the discussion that follows, the gene name nomenclature will be followed in which human genes are designated by having all letters in uppercase (e.g., SHH for Sonic Hedgehog) and their animal counterparts have only the first letter in uppercase (e.g., Shh). While the two counterparts may be considered interchangeable, this distinction serves as a reminder that not all molecular mechanisms for limb development may be conserved between humans and the mouse, chick, or other model species.
Molecular Events of the Developing Upper Limb
Molecular signaling pathways control the growth and tissue differentiation, leading to the gross anatomic milestones of limb development. These molecular events can be divided into three phases. The first phase is early limb development, which includes initial establishment of limb identity and the initiation of limb bud outgrowth. The second phase is generally considered “classical” limb development, characterized by basic patterning of the developing limb. During this phase, limb patterning is commonly subdivided into three spatial axes: proximodistal, anteroposterior (radioulnar), and dorsoventral. The third phase is characterized by growth to increase limb size and cellular differentiation to form discrete tissues that make up the individual structures of the limb.
These phases are continuous and overlapping. Early events initiate signaling processes that trigger establishment of proper limb patterning. Appropriately patterned groups of cells subsequently undergo expansion and differentiation to form specific limb structures and tissues. While it is more practical to consider each of these processes as distinct steps, these processes overlap in time and space, are dynamic, and are often interdependent through the cross talk of different signaling pathways. Disruption in any number of these events produces many of the congenital abnormalities seen clinically.
Several key tenets apply:
1.
Tissues and structures that develop concurrently may be driven by common molecular signaling pathways, although not always. Defects in common pathways may explain constellations of symptoms that are frequently seen together.
2.
Many of the molecular mechanisms identified are derived from experimental work performed in nonhuman species, especially the chicken and the mouse. These findings may or may not be conserved in human limb development.
3.
Many molecular pathways are critical for development of other organ systems. Severe defects in these pathways may not be encountered clinically due to the failure of development of major organs, resulting in a nonviable fetus. Conversely, milder defects may be encountered clinically, but necessitate screening for dysfunction in these other organ systems.
4.
Not all congenital limb differences are directly caused by changes in molecular signaling pathways. Amniotic band syndrome, for example, results from mechanical insults to normally developing tissue in utero.
5.
Specific terminology is used to distinguish clinical phenotypes due to genetic defects from nongenetic insults. This terminology is defined in Table 2.
Table 2
Terminology for etiology-based description of congenital birth defects
Terminology | Definition |
|---|---|
Sequence | A set of defects in which the steps involved in pathogenesis to produce a distinctive phenotype are known, e.g., Potter sequence |
Syndrome | Recurring pattern or constellation of defects, commonly due to genetic defect |
Association | Defects that occur together more commonly than would be expected by chance |
Disruption/deformation | External forces damages a normal developmental process |
Malformation | Genetic or developmental abnormality |
Dysplasia | Normal genetic programming, but aberrant tissue development |
For the purposes of this discussion, an exhaustive review of all the molecular events contributing to the developing limb is beyond the scope of this chapter or the need of the pediatric upper limb surgeon. Molecular events presented in this review will be key signals that play a major role in limb development and/or are clinically relevant. Two things are important for the pediatric upper limb surgeon: (1) the recognition of associated patterns of deformities and (2) the recognition of novel abnormalities or unique variants that are inheritable. The former will help with diagnosis of comorbid conditions that may help our patients, whereas the latter will help our scientist colleagues who continue deciphering the molecular mechanisms underlying the deformities that afflict our patients.
First Phase: Early Development and Limb Identity
Limb buds initiate in the region of the developing somites and first appear in the region of the upper limb on approximately day 24. This is followed 4 days later with the appearance of the lower limb buds on day 28.
Upper and lower limb bud identities are believed to be predetermined early on during cranial-caudal patterning by a conserved, sequential genetic program encoded by Hox genes (Cohn et al. 1997). Differential Hox gene expression establishes the upper limb- and lower limb-forming regions of the corresponding lateral plate mesoderm and somites from which the limb buds initiate.
These limb-forming regions dictate the morphology of the limb created in this region. Ectopic induction of the lateral plate mesoderm using Fgf-soaked beads in either region results in formation of complete upper or lower limbs depending on where the limb was initiated (Cohn et al. 1995). Determination of whether the limb develops upper or lower limb structures was correlated with proximity of bead placement to the limb-forming regions. Beads placed close to the lower limb-forming region produced ectopic lower limbs, whereas beads placed close to the upper limb-forming region induced formation of ectopic upper limbs (Cohn et al. 1995, 1997). Beads placed in between the two regions produced chimeric limbs with both upper and lower limb characteristics (Ohuchi et al. 1998).
A triggering signal is required to initiate limb bud outgrowth from the programmed limb bud forming regions of the embryo. The exact sequence of events leading to activation of growth remains unclear, but at least three transcription factors are critical for initiation of the correct upper or lower limb bud development: Tbx4, Tbx5, and Pitx1 (Agarwal et al. 2003; Duboc and Logan 2011a; Rallis et al. 2003). Tbx5 is specifically expressed in the upper limb bud, whereas Pitx1 and Tbx4 (downstream of Pitx1) are specifically expressed in the lower limb bud. The expression of these genes is quite specific and clearly identifies the limb based on this pattern in both mouse and chick embryos. Further supporting the specificity of Tbx4, Tbx5, and Pitx1, Fgf-soaked bead-induced ectopic upper limbs express Tbx5 whereas ectopic lower limbs express Tbx4 and Pitx1. Limb buds with a mixture of upper and lower limb characteristics express all three. In the upper limb, retinoic acid from the trunk plays a permissive role in limb bud initiation by permitting induction of Tbx5 expression (Cunningham et al. 2013).
Functionally, Pitx1 is clearly a determinant of lower limb morphology whereas the exact contribution of Tbx4 and Tbx5 to the development of the corresponding limb morphology is less clear. Loss of Pitx1 results in loss of lower limb characteristics, which can be rescued by Tbx4 since Tbx4 is downstream of Pitx1 signaling (Ouimette et al. 2010). Misexpression of Pitx1 in the upper limb causes a partial forelimb-to-hind limb transformation in mice and humans (Liebenberg syndrome, Mendelian Inheritance in Man [MIM] number 186550) (Spielmann et al. 2012), reflecting the role of Pitx1 in directing development of lower limb structures. Conversely, ectopic expression of Tbx4 in the upper limb does not produce the same effect. In fact, Tbx4 misexpression in the upper limb bud can substitute for Tbx5 in a conditional knockout mouse with Tbx5 deleted in the upper limb-forming region to form an intact upper limb (Minguillon et al. 2005). The Tbx5 conditional knockout mouse also indicated that Tbx5 (or misexpressed Tbx4) is needed for limb bud initiation , the absence of which results in absent upper limbs (Rallis et al. 2003).
Mutations to all three genes are associated with clinical syndromes in humans. Tbx4 mutations cause small patella syndrome (MIM 147891), characterized by patellar and hip defects. Tbx5 mutations can cause Holt-Oram syndrome (MIM 142900), characterized by upper limb and cardiac defects. Pitx1 mutations can cause clubfoot (MIM 199800) or Liebenberg syndrome (MIM 186550).
A key milestone for limb bud initiation by Tbx5 (upper limb) and Tbx4 (lower limb) is the induction of Fgf10 expression. Maintained Fgf10 expression is required for successful completion of limb bud initiation and subsequent limb development (Duboc and Logan 2011b). In the upper limb, Tbx5 is a direct activator of Fgf10 signaling (Agarwal et al. 2003). In the lower limb, some Fgf10 expression persists despite loss of Tbx4, likely due to other contributors. This overlap in signaling may explain the formed but significantly smaller lower limbs in mice with loss of Tbx4 (Naiche and Papaioannou 2003). In mice, complete loss of Fgf10 resulted in initiated limb buds but no limb outgrowth (Sekine et al. 1999). Once initiated, similar Fgf-dependent signaling mechanisms enable limb outgrowth for both upper and lower limbs.
Second Phase: Limb Patterning and Initial Growth
After limb bud initiation, outgrowth of the limb bud begins. All limb buds consist of mesodermal tissue originating from lateral plate mesoderm (forms bone, cartilage, and tendons) and somitic mesoderm (forms muscles, nerves, and vasculature) and are covered by a layer of ectoderm. Anatomically, the limb goes through a progressive set of morphological changes starting with a thin nubbin of tissue along the chest wall of the embryo through the limb paddle stages. During this process, limb development can be thought of as occurring in three spatially distinct axes. Each of these limb growth axes contains a signaling center, an area or group of cells, that is responsible for establishing the corresponding axis. The three axes are proximodistal (PD), anteroposterior (AP) (radioulnar or pre-/postaxial), and dorsoventral (DV).
Proximodistal (PD)
At the distal tip of the developing limb buds , at the interface between dorsal and ventral ectoderm, a ridge of thickened ectoderm forms in response to signals from the underlying mesoderm. This thickening develops into the apical ectodermal ridge (AER), the signaling center for the PD axis (Fig. 2). Signaling from the AER is critical for limb bud outgrowth and elongation, directly inducing formation of upper limb structures in a proximal (early) to distal (late) manner. These structures may be viewed as three distinct segments achieved by patterning along the proximodistal axis: the stylopod (upper arm), zeugopod (forearm), and autopod (wrist and hand). This PD patterning appears to occur very early when the AER is established in the initiating limb bud and is dependent on FGF signaling from the AER (Mariani et al. 2008; Sun et al. 2002).
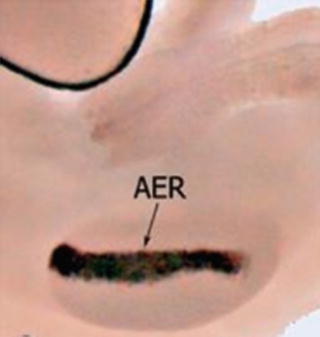
Fig. 2
Whole-mount RNA in situ hybridization visualization the AER at the tip of a developing mouse limb bud (Soshnikova and Birchmeier 2006)
The importance of the AER is demonstrated by limb differences along the PD axis if AER function is impaired. Disruption results in limb truncations at a level corresponding with the stage of development when the disruption occurred (Summerbell 1974). The later the removal or disruption of the apical ectodermal ridge in development, the more distal the resulting truncation. Structures proximal to the level of truncation remain intact. These truncations can be rescued by grafting an AER from a different chick embryo, defining the AER as both necessary and sufficient to promote outgrowth.
The molecular cue from the AER was isolated and identified to be one of several growth factors from the fibroblast growth factor (Fgf) family. In the developing limb, members of the Fgf family exhibit varying degrees of functional redundancy. Of the Fgfs, Fgf10 alone is both necessary and sufficient to produce an intact limb (Duboc and Logan 2011b) and is normally expressed by the mesenchyme underlying the AER. The AER itself specifically expresses Fgf4, Fgf8, Fgf9, and Fgf17 (the AER-FGFs). Mice deficient in Fgf4, Fgf9, or Fgf17 retain normal limb development, likely rescued by the functional redundancy of the Fgfs. In contrast, Fgf8 is critical as loss of Fgf8 caused impaired limb outgrowth and significantly smaller limbs (Mariani et al. 2008).
Fgf10 from the sub-AER mesenchyme and Fgf8 from the AER are intrinsically related in a positive feedback loop that forms the core signaling required for growth along the PD axis (Ohuchi et al. 1997). Early expression of Tbx5 during upper limb bud initiation first induces expression of Fgf10 from the sub-AER mesenchyme (Agarwal et al. 2003). Fgf10 signals to the overlying AER to express Wnt3a, which in turn drives Fgf8 expression. Fgf8 from the AER then signals to the sub-AER mesenchyme to maintain Fgf10 expression from the underlying mesenchyme, resulting in an Fgf8-Fgf10 positive feedback loop located at the distal end of the developing limb. This Fgf/Wnt signaling loop is critical for limb development – loss of Wnt3 (tetra-amelia, MIM 273395) (Niemann et al. 2004) or Fgf10 (Sekine et al. 1999) results in amelia, or absence of limb formation.
The importance of the distal end of the limb bud in limb outgrowth was initially conceptualized in a “progress zone” model to explain proximodistal development. The progress zone model posits that there is a zone of undifferentiated mesenchymal cells within the sub-AER mesenchyme with an intrinsic timing mechanism. The cells in this zone are maintained by Wnt3a and Fgf8 from the AER, which maintains a pool of progenitor cells by stimulating proliferation and inhibiting differentiation (ten Berge et al. 2008). A timing mechanism would provide progenitor cells with positional cues based on how long the cells reside in the progress zone. Mesenchymal stem cells that exit relatively early develop into proximal structures such as the humerus. Cells that exit late end up in distal locations as the limb elongates and develop into distal structures such as the forearm and hand. The time-dependent mechanism of the progress zone model provided a mechanistic explanation for the time-dependent transverse deficit phenotype produced by removal of the AER. The later the AER excision, the more distal the defects due to loss of progenitors with longer residence in the progress zone.
Subsequent experiments, however, demonstrated that the progress zone model is inaccurate. Lineage tracing in X-irradiation experiments to induce phocomelia in chick embryos demonstrated that proximodistal patterning was unaffected despite radiation-induced defects in proximal structures (Galloway et al. 2009). Similarly, fate mapping in chick limb buds demonstrated that proximodistal cell fates were established early, with limb truncations occurring due to apoptosis of these fated cells rather than defects in PD patterning (Dudley et al. 2002).
Modern attempts to understand PD patterning emphasizes the conceptualization of limb patterning in the context of dynamic interactions between molecular events (Tabin and Wolpert 2007). More recently, a “two-signal” model was proposed to explain proximodistal determination of limb structures. Specifically, the two opposing signals were retinoic acid (RA) for induction of proximal structures, with the FGFs from the AER determining formation of distal structures (Cooper et al. 2011; Roselló-Díez et al. 2011). Supporting this model, ectopic introduction of RA to the distal limb bud resulted in proximalization of the distal limb (Mercader et al. 2000). Additionally, the AER-FGFs were demonstrated to establish distal structures by repression of Meis1/2, homeobox genes that establish the proximal limb (Mariani et al. 2008).
The role of retinoic acid appears to be permissive rather than actively establishing PD patterning (Cunningham et al. 2013). Fgf8 expression by the developing heart suppresses limb bud initiation due to inhibition of Tbx5 expression. Retinoic acid from the trunk functions to block cardiac Fgf8, enabling limb bud expression of Tbx5 and Meis1/2 and limb bud initiation. For proximodistal patterning itself, RA is unnecessary. Interestingly, Tbx5 itself is needed for cardiomyocyte differentiation (Holt-Oram syndrome, MIM 142900), the mutation of which is characterized by upper limb and cardiac defects.
PD patterning remains incompletely understood and is an evolving field. It is also not known the extent to which these mechanisms are conserved in human limb development.
Despite our lack of understanding, the clinical implications have not changed. Defects along the proximodistal axis, such as limb truncations and longitudinal defects, remain intrinsically related to the AER and FGF function. Insult to the AER during development will lead to clinically observed truncations at variable stages depending on the timing of the insult. Frequently, isolated limb cases suspected to be secondary to insults to the AER will predominantly be mechanical and nonheritable due to the extensive role FGFs play in other biological systems, resulting in severe systemic defects that may render limb defects lower in priority.
Anteroposterior (AP)
Arguably, the most studied axis of limb patterning, and perhaps the most clinically relevant, is the AP axis. The anteroposterior axis distinguishes the radial (or preaxial) from the ulnar (or postaxial) side. This axis was initially discovered when sections of posterior (ulnar) limb bud tissue were excised at varying stages of chicken limb development resulting in limbs that developed longitudinally but that did not develop radioulnar-based digit identities. Excision of radial tissue did not produce the same effect. It was discovered that a small region of posterior cells, at the junction of the limb paddle and the trunk, was a signaling center for AP development termed the zone of polarizing activity (ZPA). This zone of tissue polarized the limb along the AP axis. When ZPA tissue was grafted onto the radial side, a mirror image of the limb along the AP plane was produced (Tickle 1981).
The key signaling molecule of the ZPA is Sonic Hedgehog (SHH). This ligand was named for its molecular resemblance to the drosophila molecule Hedgehog, which is important for fly segmentation, thus leading to its similar name despite different phyla. SHH is a diffusible signaling molecule with expression restricted to the posteriorly located ZPA (Fig. 3) and forms a posterior (high) to anterior (low) gradient of SHH signaling. Functionally, SHH is critical for regulating AP patterning and growth of the zeugopod (forearm) and autopod (hand).
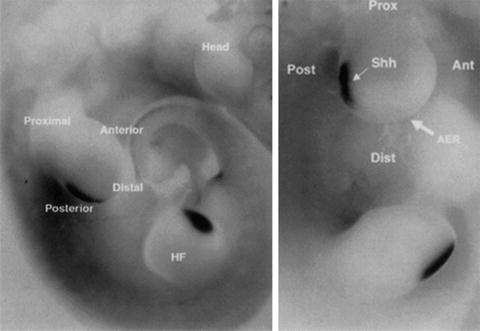
Fig. 3
In situ hybridization for SHH, visualizing the SHH-expressing ZPA located in the posterior margin of both the upper and lower limb buds in a mouse embryo (e10.5) (Daluiski et al. 2001)
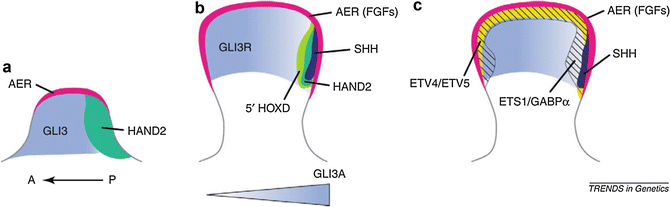
Restriction of SHH expression to the posterior-located ZPA is accomplished by Gli3, a transcription factor that pre-patterns the early limb bud along the AP axis before SHH expression is activated (te Welscher et al. 2002). Gli3 exists in two forms: GLI3A (activator) and GLI3R (repressor). By default, Gli3 is modified to form the Gli3 repressor form. On the posterior (ZPA) side, SHH inhibits this modification to allow for production of the GLI3A activator form, producing a gradient of high GLI3A:GLI3R ratio posteriorly to low GLI3A:GLI3R anteriorly. Manipulation in chick embryos to produce high GLI3A:GLI3R ratios throughout results in polydactyly with posterior digit identities of all extraneous digits (Litingtung et al. 2002), reflecting the role of high SHH signaling in specifying posterior structures. Of note, the stylopod (upper arm) is patterned independently of SHH, presumably accomplished by Gli3-mediated pre-patterning (Niswander 2002). The elbow represents the transition from SHH-independent to SHH-dependent limb development, which may have clinical implications in such clinical phenotypes as below-elbow truncations.
The interactions between SHH and Gli3 result in a gradient of SHH signaling and GLI3A:GLI3R ratio along the AP axis that directs specification and development of autopod ulnar-sided to radial-sided structures (Fig. 4). In humans, defects in the function of either SHH or Gli3 may cause limb defects along the AP axis, frequently manifesting clinically with digit abnormalities such as polydactyly and syndactyly (Anderson et al. 2012). Mutations of GLI3, for example, result in various preaxial and postaxial polydactylies in Greig cephalopolysyndactyly syndrome (MIM 175700) (Hui and Joyner 1993).
Defects in digit development are closely correlated with SHH function because SHH signaling specifies the number of digits as well as the identity of each digit. To achieve this, SHH is expressed in a gradient that varies both spatially and temporally (Harfe et al. 2004). Spatially, the concentration gradient exposes the mesoderm to varying concentrations of SHH, with the concentrations greatest on the posterior (ulnar) side and none on the anterior (radial) side. The mesenchymal progenitors of the fifth digit are exposed to the greatest SHH concentration whereas the thumb develops in the absence of SHH signaling. Temporally, the length of exposure to SHH determines digit identity. Shorter-term exposure of the mesenchyme is sufficient to specify anterior digits (second digit), whereas the fifth digit requires the longest SHH exposure for correct specification (Scherz et al. 2007). In fact, the posterior three digits contain SHH-expressing cells from the ZPA while the SHH contribution to the second digit derives from paracrine signaling (Harfe et al. 2004). Furthermore, SHH acts as a mitogen to produce the necessary progenitor pool for forming five complete digits (Malik 2014).
Digit identity is established by the SHH gradient from the ZPA , but digits initially develop as “webbed” fingers. Extraneous tissue between fingers must undergo apoptosis to form digits with interdigital separations. Bone morphogenetic proteins (BMPs), widely known for their role in chondrogenesis and osteogenesis, mediate apoptosis of the interdigital mesenchyme to produce separated fingers. The interdigital mesenchyme expresses BMP2, BMP4, and BMP7 which antagonizes the pro-survival effects of the Fgfs from the overlying AER (Pajni-Underwood et al. 2007; Suzuki 2013). In animals with webbed limbs such as bats, BMP antagonists block BMP signaling to produce persistent interdigital tissue (Oberg et al. 2010). Enhanced function or signaling of Fgfs can also prevent interdigital apoptosis by overcoming BMP-mediated inhibition, as occurs in the syndactyly observed in Apert syndrome (MIM 101200).
The factors governing formation of the digits are less well understood. Digit formation is currently thought to occur from the combination of the initial AP patterning established by SHH, BMP signaling from the interdigital mesenchyme, and phalangeal growth mediated by the phalanx-forming region (PFR) found in the sub-AER mesenchyme. Cells from the sub-AER mesenchyme are continuously incorporated into each of the PFRs and subsequently into the growing digit. Cells incorporated earlier form the proximal digit, undergoing condensation to form the proximal phalanges; cells incorporated later form the distal phalanges. BMP signaling from the interdigital mesenchyme promotes this process via its stimulatory effects on the PFR, in contrast to the inhibitory effect BMPs have on the AER-FGFs that produces interdigital apoptosis (Suzuki et al. 2008).
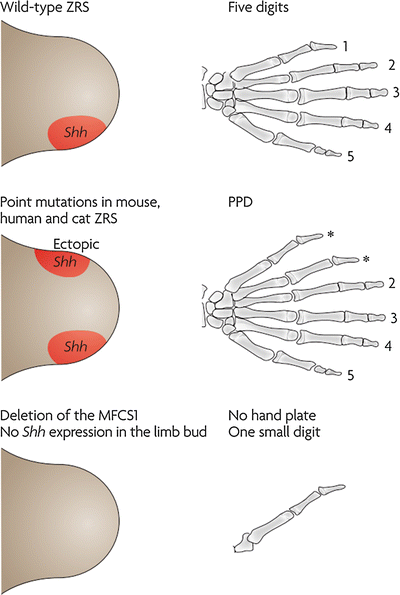
SHH is critical in various development processes, most notably is the development of the notochord. Mutations to the SHH gene proper, such as large deletions that produce a defective protein, occur but are unlikely to be encountered, due to either developmental lethality or serious neurologic defects that take precedence. More relevant, however, are mutations to regulators of SHH signaling. Several of these regulators have clinical importance and include ZRS mutations, GLI3 mutations, and ciliopathies.
The ZRS (ZPA regulatory sequence) is an enhancer sequence that is necessary and sufficient for regulating the spatiotemporal SHH activity in the developing limb (Anderson et al. 2012). This sequence is located at a distance (1 Mb upstream) from SHH, in an intron in the LMBR1 gene. Mutations to this region produces a range of phenotypes involving the upper limb including preaxial polydactyly (MIM 174500), isolated triphalangeal thumb (MIM 174500), syndromic triphalangeal thumb (MIM 174500), syndactyly (MIM 186200), and acheiropody (bilateral congenital amputations, MIM 200500). Triphalangeal thumbs and preaxial polydactyly arise secondary to point mutations that result in ectopic SHH expression, producing an ectopic ZPA at the anterior margin of the limb bud (Lettice et al. 2008). This ectopic SHH may respecify anterior digits into posterior digits (triphalangeal thumb or thumb-to-finger transformation) or induce the formation of a mirror hand (duplication of posterior digits in the anterior margin) (Fig. 5). Syndactyly and polysyndactyly may occur from overexpression of SHH, particularly in the interdigital mesenchyme, and are associated with mutations that increase the dosage of SHH such as ZRS duplications (Klopocki et al. 2008) or the adoption of a more widely expressed enhancer by SHH (Anderson et al. 2012). Acheiropodia, characterized by congenital upper and lower limb amputations and aplasia of the hands and feet, is associated with mutations to the LMBR1 gene not involving the ZRS sequence, raising the possibility of additional regulatory sites in addition to the ZRS (Ianakiev et al. 2001).