Chapter 102 Dysmorphology
Dysmorphology is the study of abnormalities of human form and the mechanisms that cause them. It is estimated that 1 in 40, or 2.5% of newborns, have a recognizable malformation or malformations at birth. In about half of these newborns, a single isolated malformation is found, whereas in the other half there are multiple malformations. It is estimated that 10% of pediatric hospital admissions involve known genetic conditions, 18% involve congenital defects of unknown etiology, and 40% of surgical admissions are of patients with congenital malformations. Twenty percent to 30% of infant deaths and 30-50% of deaths after the neonatal period are due to congenital abnormalities (http://www.marchofdimes.com/peristats/). In 2001, birth defects accounted for 1 in 5 infant deaths in the United States, with a rate of 137.6 deaths per 100,000 live births, which is higher than other causes such as preterm/low birthweight (109.5/100,000), sudden infant death syndrome (55.5/100,000), maternal complications of pregnancy (37.3/100,000), and respiratory distress syndrome (25.3/100,000).
Classification of Birth Defects
Congenital birth defects either are isolated, single defects or manifest as multiple anomalies in a single individual. Single primary defects can be classified according to the nature of the presumed cause of the defect as a malformation, dysplasia, deformation, or disruption (Table 102-1). Malformations and dysplasias both affect intrinsic structure. A malformation is a primary structural defect arising from a localized error in morphogenesis and resulting in the abnormal formation of a tissue or organ. Dysplasia refers to an abnormal organization of cells into tissues. The distinction of a malformation from a dysplasia may be helpful, but there is much overlap. Deformations and disruptions are secondary effects that result from forces generated extrinsic to the affected tissue or organ. A deformation is an alteration in shape or structure of a structure or organ that has differentiated normally. A disruption is a structural defect resulting from the destruction of a structure that had formed normally before the insult.
Table 102-1 MECHANISMS, TERMINOLOGY, AND DEFINITIONS OF DYSMORPHOLOGY
| TERMINOLOGY | DEFINITION | EXAMPLE |
|---|---|---|
| Malformation sequence | Single, local tissue morphogenesis abnormality that produces a chain of subsequent defects | DiGeorge sequence of primary fourth brachial arch and 3rd and 4th pharyngeal pouch defects that lead to aplasia or hypoplasia of the thymus and parathyroid glands, aortic arch anomalies, and micrognathia |
| Deformation sequence | Mechanical (uterine) forces that alter structure of intrinsically normal tissue | Oligohydramnios produces deformations by in utero compression of limbs (dislocated hips, equinovarus foot deformity), crumpled ears, dislocated nose, or small thorax |
| Disruption sequence | In utero tissue destruction after a period of normal morphogenesis | Amnionic membrane rupture sequence, leading to amputation of fingers/toes, tissue fibrosis, and destructive tissue bands |
| Dysplasia sequence | Poor organization of cells into tissues or organs | Neurocutaneous melanosis sequence with poor migration of melanocyte precursor cells from the neural crest to the periphery, manifesting as melanocytic hamartosis of skin, meninges, and so forth |
| Malformation syndrome | Appearance of multiple malformations in unrelated tissues without an understandable unifying cause; with enhanced genetic investigation, a single etiology may become identified |
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier Saunders.
Malformations and/or Dysplasias
Human malformations and dysplasias are caused by the combined effects of genes and environmental factors (Table 102-2). Some malformations are caused by single gene defects or abnormalities of multiple genes acting in concert, and the environment causes others. In 1996, it was thought that malformations were due to monogenic defects in 7.5% of patients; to chromosomal anomalies in 6%; to multigenic defects in 20%; and to known environmental factors, such as maternal diseases, infections, and teratogens, in 6-7% (Table 102-3); in the remaining 60-70% of patients, malformations were classified as due to unknown etiologies. A decade later, the percentages were somewhat higher for all categories of known causes of malformations, a change resulting from improved cytogenetic methods of detecting small chromosomal abnormalities as well as techniques for mapping and cloning disease genes. Since the previous edition of this book, an additional 10-20% of birth defects result from even smaller chromosomal abnormalities detectable by whole genome arrays using comparative genomic hybridization (array CGH) methods. In spite of these advances, we still do not know the causes or even the diagnosis for 40-50% of birth defects.
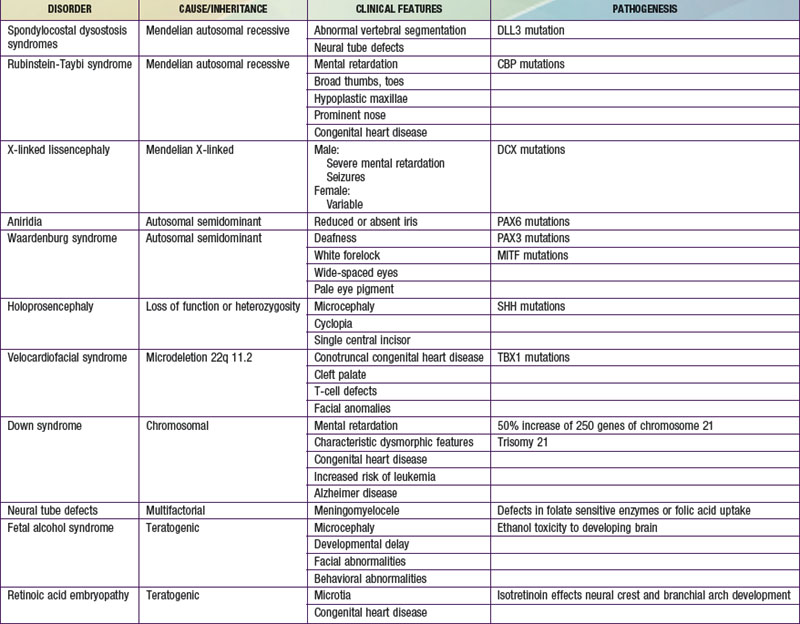
Table 102-3 CAUSES OF CONGENITAL MALFORMATIONS
MONOGENIC (7.5% of serious anomalies)
CHROMOSOMAL (6% of serious anomalies)
MATERNAL INFECTION (2% of serious anomalies)
Intrauterine infections (e.g., herpes simplex virus, cytomegalovirus, varicella-zoster virus, rubella virus, and toxoplasmosis)
MATERNAL ILLNESS (3.5% of serious anomalies)
UTERINE ENVIRONMENT (% unknown)
ENVIRONMENTAL AGENTS (% unknown)
MEDICATIONS (% unknown)
UNKNOWN ETIOLOGIES
SPORADIC SYNDROME COMPLEXES (ANOMALADS)
NUTRITIONAL
Low folic acid–neural tube defects
From Behrman RE, Kliegman RM, editors: Nelson’s essentials of pediatrics, ed 4, Philadelphia, 2002, WB Saunders.
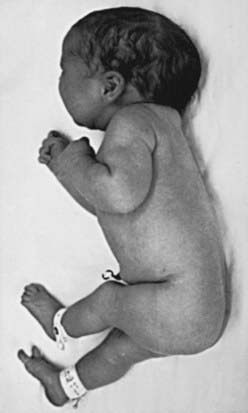
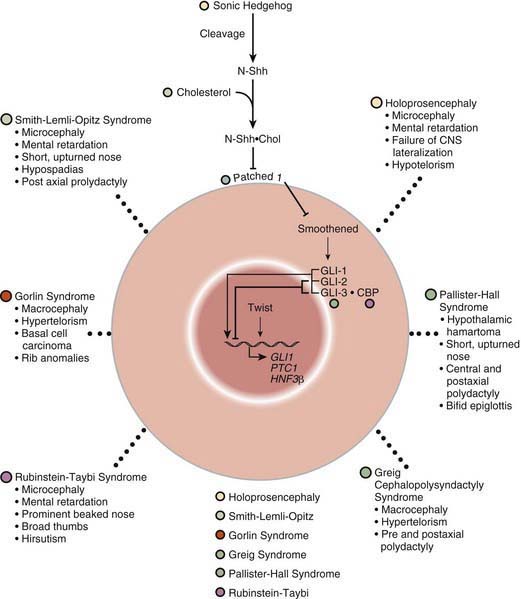
Many developmental abnormalities are caused by mutations in a single gene and display characteristic mendelian patterns of inheritance. The molecular etiology for more than 250 single gene disorders is known. Affected genes are often part of evolutionarily conserved signal transduction pathways, transcription factors, or regulatory proteins required for key developmental events. Some examples are listed in Table 102-2; they include autosomal recessive spondylocostal dysostosis (SCD) syndrome, the autosomal recessive Smith-Lemli-Opitz syndrome (SLOS), the autosomal dominant Rubinstein-Taybi syndrome, and the X-linked lissencephaly (“smooth brain”) syndrome. The SCD syndrome is etiologically heterogeneous and is often caused by mutations in the gene coding for delta-like 3 (DLL3), a ligand of the Notch receptors. The Notch/delta pathway is conserved throughout evolution and regulates a number of developmental events. Patients with SCD display a characteristic pattern of abnormal vertebral segmentation associated with a number of other malformations, such as neural tube defects. SLOS (Fig. 102-1) results from mutations in the sterol delta-7-reductase gene, an enzyme important in cholesterol biosynthesis. Patients with SLOS display syndactyly (fusion of the fingers and toes) often with polydactyly, upturned nose, ptosis, cryptorchidism, central nervous system hypoplasia, and holoprosencephaly. These mutations link cholesterol biosynthesis pathogenetically to the sonic hedgehog (SHH) pathway, because many of the features of the former disorder are related to defects in SHH, which is post-translationally modified by cholesterol (Chapter 80). Rubinstein-Taybi syndrome (see Fig. 102-1) results from heterozygous loss-of-function mutations in the gene coding for a broadly acting transcriptional coactivator called CBP, or CREB-binding protein. The CBP coactivator regulates the transcription of a number of genes, a fact that helps explain why patients with mutations in CBP have a wide-ranging phenotype that includes mental retardation, broad thumbs and toes, and congenital heart disease. One of the transcription factors that binds to CBP is GLI3, a transcription factor that is part of the SHH pathway (see Fig. 102-1). X-linked lissencephaly—a severe neuronal migration defect that in males causes a smooth brain with reduction or absence of gyri and sulci and in females gives rise to a variable pattern of mental retardation and seizures—is caused by mutations in DCX, a protein that regulates the activity of dynein motors and moves the nucleus during neuronal migration.
Other malformation syndromes are caused by chromosomal imbalance, multifactorial inheritance, and teratogens (see Tables 102-2 and 102-3). Down syndrome results from an extra dose of part or all of chromosome 21, a small chromosome that contains ≈ 200 known or predicted genes. It is most commonly caused by trisomy 21, which means that individuals with Down syndrome have an increased dose of as many as 250 genes contained on this chromosome (Chapter 76.1). Neural tube defects (NTDs) are an example of a disorder that displays multifactorial inheritance in the majority of cases. NTDs and a number of other congenital malformations, such as cleft lip and palate, recur in families, but several genes and environmental factors together contribute to the pathogenesis (see Table 102-2). Many of the genes involved in NTDs are unknown, so one cannot predict with certainty a mode of inheritance or a precise recurrence risk. Empiric risks can be provided on the basis of population studies and the presence of single or multiple relatives with the same malformation. However, an important gene/environment interaction has been identified for NTDs (Chapter 585.1). Folic acid status is associated with NTDs and can result from a combination of dietary deficiencies and increased utilization during pregnancy as well as from a common variant in the gene for an enzyme in the folate recycling pathway, 5,10-methylene-tetrahydrofolate reductase, that makes this enzyme less stable. These discoveries led to the recommendation that all women supplement their diets with 400-800 µg of folic acid per day 1 mo before pregnancy and during the 1st 2 mo of pregnancy. This supplementation has resulted in a reduction in the incidence of NTDs by 75%. Several teratogenic causes of birth defects have been described (see Tables 102-2 and 102-3). Ethanol causes a recognizable malformation syndrome called fetal alcohol syndrome (FAS) (Chapter 100.2). Children with FAS display microcephaly, developmental delay, hyperactivity, and facial dysmorphisms. Ethanol, which is toxic to the developing central nervous system, causes cell death of developing neurons.
Deformations
Most deformations involve the musculoskeletal system (Fig. 102-2). Fetal movement is required for the proper development of the normal musculoskeletal system, and anything that restricts fetal movement can cause a musculoskeletal deformation from intrauterine molding. It is important to recognize that deformations can be caused by problems either intrinsic or extrinsic to the developing fetus. Two major intrinsic causes of deformations are primary neuromuscular disorders and oligohydramnios, or decreased amniotic fluid, which is caused by renal defects. The major extrinsic causes of deformation are those that result in fetal crowding to restrict fetal movement. Examples of such extrinsic causes are oligohydramnios from chronic leakage of amniotic fluid, breech presentation (Fig. 102-3), and abnormal shape of the amniotic cavity. When a fetus is in the breech position, the incidence of deformations is increased 10-fold. The shape of the amniotic cavity has a profound effect on the shape of the fetus and is influenced by many factors, including uterine shape; volume of amniotic fluid; size and shape of the fetus; presence of more than one fetus; site of placental implantation; presence of uterine tumors; shape of the abdominal cavity, which is influenced by the pelvis, sacral promontory, and neighboring abdominal organs; and tightness of the abdominal musculature.
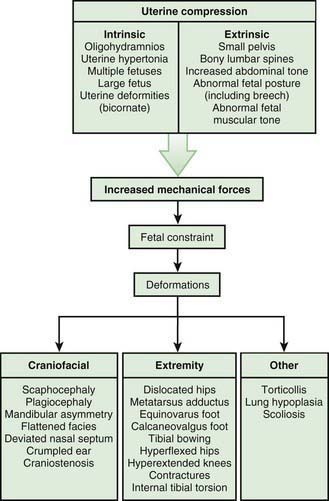
Figure 102-2 Deformation abnormalities resulting from uterine compression.
(From Kliegman RM, Jenson HB, Marcdante KJ, et al, editors: Nelson essentials of pediatrics, ed 5, Philadelphia, 2005, Saunders.)