Drugs for Hyperbilirubinemia
David K. Stevenson
Ronald J. Wong
Susan R. Hintz
Hendrik J. Vreman
Introduction
Hyperbilirubinemia is a natural and essentially ubiquitous transitional phenomenon among human newborns. Approximately 60% to 70% of all term infants, and nearly all premature infants, become visibly jaundiced during the first week of life after birth. For term infants, the serum or plasma total bilirubin (PTB) concentration typically peaks 3 to 4 days after birth in the range of 5 to 6 mg per dL (86 to 103 μmol per L). For premature infants, PTB levels peak later and higher after the first several days of life. Although much uncertainty and debate remains concerning the range of PTB considered as benign physiologic jaundice, the consensus places the maximal “safe” peak PTB threshold at approximately 17 mg per dL (291 μmol per L) for otherwise healthy term and late preterm babies. Because hyperbilirubinemia above this threshold is considered to be pathologic, the etiology of the hyperbilirubinemia should be investigated and appropriate therapy considered or initiated depending on the clinical circumstances (1). In immature infants, treatment to decrease PTB levels is often initiated at lower PTB levels because of lower albumin levels and diminished affinity of albumin for bilirubin in these infants. Moreover, binding to albumin is least avid in the early transitional period after birth and is influenced adversely by any confounding conditions, such as infection or acidosis, that increase free bilirubin in circulation and the likelihood of movement into tissues.
Historically, the main therapies for neonatal hyperbilirubinemia have been phototherapy and exchange transfusion. Light could be considered a drug for hyperbilirubinemia, but most physicians pay little attention to the radiometric qualities (effective spectral width and peak emission) and quantities [irradiance (μW per cm2 per nm)] involved, or the factors that affect the dose of phototherapy (duration, body surface area exposure) (2,3). Finally, issues that are involved in producing phototherapy-related side effects (riboflavin destruction, erythema, and photosensitizing drugs) also deserve attention. A discussion of these topics should include reference to the qualities of light-emitting diodes because of their high intensity and narrowband light in the spectrum of choice with minimal heat generation (3,4,5,6), but these issues are beyond the scope of this chapter. In spite of the proven benefits of phototherapy and maximal spatial limitations, the understanding of the biology of newborn jaundice and the existence and further development of alternative pharmacologic therapies for neonatal hyperbilirubinemia are an integral part of the management of neonatal jaundice and its consequences.
Neonatal Jaundice
Neonatal jaundice is the result of an imbalance between the production of bilirubin and its elimination (7,8,9). Bilirubin production on a body weight basis is increased in the newborn by approximately two to three times that of an adult (10,11). This relative increase in bilirubin production in the newborn is the result of an increased circulating red cell mass and a shortened red cell life span. Consequently, all newborn infants have increased bilirubin production as a contributing cause of their transitional or pathologic jaundice. The pattern of hyperbilirubinemia (its peak and duration) is influenced further by the efficiency with which the pigment is eliminated. The major factor contributing to impaired elimination in the transitional period after birth is decreased hepatic conjugation of bilirubin. The gradual induction of uridine diphosphoglucuronate glucuronosyltransferase (UGT) contributes most importantly to the pattern of hyperbilirubinemia after birth because changes in bilirubin production are slower and more gradual within the time frame of the rapid elevation in PTB levels after birth and the decline in the latter part of the first week and into the second week of life. Thus, because all newborn babies have temporarily impaired conjugation, any pathologic state associated with increased bilirubin production, such as hemolysis, represents a serious risk to the newborn infant, especially in the first several days of life. Even without pathologic elevations in bilirubin production, greater impairments in conjugation associated with conditions such as Gilbert syndrome (12,13,14) and the Asian G71R mutation in the UGT1A1 gene (14,15,16) can place infants at risk for kernicterus because of unexpected alterations in the pattern of hyperbilirubinemia, including its peak and duration. In particular, the coexpression of gene polymorphisms involved in bilirubin production, such as (GT)n, repeats in the HO-1 promoter and glucose-6-phosphate dehydrogenase
(G6PD) mutations, and metabolisms, such as OATP1A1 and UGT1A1 and the TATAA box variants, may provide genetic markers for clinical risk assessments, as well as potential therapeutic targets (17,18).
(G6PD) mutations, and metabolisms, such as OATP1A1 and UGT1A1 and the TATAA box variants, may provide genetic markers for clinical risk assessments, as well as potential therapeutic targets (17,18).
The imbalances in the production of the pigment and its elimination have been well studied, and various methods have been proposed to identify infants at risk for severe hyperbilirubinemia. Because the predominant source of carbon monoxide (CO) in the body is the degradation of heme, which ultimately leads to the production of equimolar amounts of bilirubin, increased bilirubin production can be estimated by measuring the end-tidal CO in breath, or carboxyhemoglobin (COHb) in circulation, after these measurements are corrected for ambient CO (ETCOc or COHbc, respectively) (19,20). The normalization of COHbc to hemoglobin concentration (COHbc/Hb) can serve as an even more sensitive index of excessive red cell destruction (21). For example, the infant with hemolytic anemia would have a higher COHbc/Hb ratio than the infant with anemia caused by blood loss. By measuring the conjugated fraction of bilirubin, another index can be applied, which assesses the relative balance between bilirubin production and conjugation [COHbc/TCB (%)], where TCB is the total conjugated bilirubin (9). Finally, a nomogram plotting hour-specific bilirubin levels is also informed by the balance of bilirubin production and elimination over time (22). Because all infants have impaired conjugation during the transitional period, deviations from the percentile tracks in the first several days of life are most often related to a relative increase in bilirubin production, whereas deviations after the first week of life are more likely the result of persistent impairment in bilirubin conjugation and therefore elimination.
The logic of removing bilirubin from circulation after it has been produced is clear, but it is also only reactionary and may not avoid potential neurologic injury in every circumstance. Another, more rational, treatment strategy would be to inhibit bilirubin production, thus ameliorating the primary contributing factor in neonatal hyperbilirubinemia and the risk for kernicterus. If a safe drug for inhibiting bilirubin production could be identified, its use could be universalized. However, another alternative preventive strategy could involve the early rapid and accurate identification of infants at risk for increased bilirubin production or at least with an imbalance between the production and the elimination of bilirubin. One direct approach would be to measure ETCOc or COHbc as an index of bilirubin formation and, thus, identify high producers of the pigment for targeted therapy. Yet another approach would be to plot hour-specific bilirubin levels and be cognizant of early deviations from the nomogram suggestive of increased bilirubin production or the combination of relatively insufficient conjugation for a given bilirubin load (23). Whether the approach is targeted would depend, at least in part, on the safety, efficacy, and cost of the chemotherapeutic agent.
Heme Degradation Pathway
Heme is degraded in a two-step enzymatic pathway, which requires molecular oxygen and NADPH (see Fig. 18.1). The first step is rate limiting and catalyzed by heme oxygenase (HO), a membrane-bound enzyme (24). In this first step, the porphyrin macrocycle is broken at the 9-α-meso carbon bridge after a series of oxidations and reductions, and liberates CO, iron, and biliverdin in equimolar amounts. In the second enzymatic step, biliverdin is immediately reduced in the cytosol by biliverdin reductase in an NADPH-dependent reaction to generate bilirubin. Because HO is the rate-limiting enzyme in the pathway, its inhibition results in decreased production of CO, iron, and biliverdin, thus leading to decreased bilirubin production (25,26).
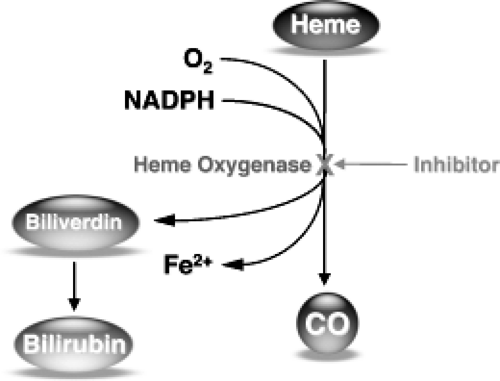 Figure 18.1. Heme catabolic pathway. |
Heme Oxygenase Inhibitors
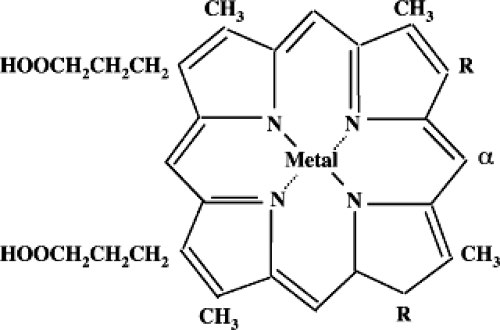 Figure 18.2. Metalloporphyrin structure. (From Vreman HJ, Wong RJ, Stevenson DK. Alternative metalloporphyrins for the treatment of neonatal jaundice. J Perinatol 2001; 21(Suppl 1):S108–S113, with permission.) |
Metalloporphyrins
Many synthetic structural analogues or metalloporphyrins (Mps) of heme (ferro-protoporphyrin) are effective in vitro and in vivo competitive inhibitors of HO (Fig. 18.2, Table 18.1). The original idea for using heme analogues (i.e., zinc protoporphyrin, ZnPP) as drugs for modulating bilirubin production was pioneered in 1981 by Maines (27) and has driven nearly three decades of intensive investigation of a variety of related potential chemopreventive agents. These compounds proved to have variable efficacies with respect to the two primary and well-described isoforms of HO, the inducible form (HO-1) and the constitutive form (HO-2), which occur in different relative proportions in tissues (28,29). Moreover, some of
these compounds have been found to also inhibit other enzymes, such as nitric oxide synthase (NOS) and soluble guanylyl cyclase (sGC), as well as processes such as lipid peroxidation (30,31). In fact, the products of heme degradation, CO, iron, biliverdin, and bilirubin all have been shown to have important biological roles as antioxidant and anti-inflammatory agents (20,32,33,34). Thus, the deliberate attenuation of heme degradation for the purpose of controlling the production of bilirubin must be considered in the context of the potential side effects on other important biologic processes in the transitional period and their effects beyond (35).
these compounds have been found to also inhibit other enzymes, such as nitric oxide synthase (NOS) and soluble guanylyl cyclase (sGC), as well as processes such as lipid peroxidation (30,31). In fact, the products of heme degradation, CO, iron, biliverdin, and bilirubin all have been shown to have important biological roles as antioxidant and anti-inflammatory agents (20,32,33,34). Thus, the deliberate attenuation of heme degradation for the purpose of controlling the production of bilirubin must be considered in the context of the potential side effects on other important biologic processes in the transitional period and their effects beyond (35).
Table 18.1 Porphyrin Type Based on Chelated Metal and Ring Substituent | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Tin protoporphyrin (SnPP) was the first synthetic heme analogue used for the purpose of inhibiting HO in human neonates, after intensive investigation in rodents and nonhuman primates (36,37,38). Although highly efficacious, the photoreactivity of this Mp made it a less desirable drug (39,40,41). Tin mesoporphyrin (SnMP), which is also photoreactive and more potent, however, has been used in several randomized controlled trials in human neonates at considerably lower doses than was possible with SnPP (42,43). In fact, a single intramuscular dose of 6 and 4.5 μM SnMP per kg body weight has been shown to eliminate the need for phototherapy (42) or for exchange transfusion (44), respectively, during the postnatal period. The efficacy of the compound has been well established, but it still may not represent the ideal therapeutic agent because it is photoreactive and contains a foreign (nonessential) metal, induces the HO-1 gene, and can inhibit other enzyme systems such as NOS and sGC. ZnPP has been proposed as an alternative drug, but its inhibitory potency is much lower and its formulation for administration has been more difficult (26). Nonetheless, it is a naturally occurring Mp and has both in vitro and in vivo inhibitory properties for both HO-1 and HO-2, and has been shown to suppress hyperbilirubinemia in neonatal rodents and nonhuman primates (40). Moreover, this naturally occurring heme analogue has no apparent photoreactivity in vivo. With respect to photoreactivity, many of the heme analogues have already been characterized along with their inhibitory potency (26,40,41,45).
In addition to screening for potential phototoxicity, the impact of these heme analogues on the induction of HO-1 is also important to consider (26,46,47,48). In fact, the various Mps also differ in their ability to upregulate the HO-1 gene. For example, ZnPP and zinc bis glycol porphyrin (ZnBG) appear to cause minimal induction. The latter compound is a very potent inhibitor, which is orally absorbable (46,47,49,50). Although ZnBG is photoreactive, its substantial potency would allow for its use with minimal clinical risk similar to what has been observed for SnMP. The characteristics of the unique metabolite ZnPP have been reviewed in detail elsewhere (51). Besides its therapeutic potential, it also has clinical applications for assessing nutritional iron status in pediatric patients, pregnant women, and blood donors and for diagnosing other disorders in iron metabolism including lead toxicity (51). With respect to the potential for inducing HO-1, preliminary data on cadmium-induced HO-1 transcription suggest a possible programming effect, with a second exposure to the drug resulting in less of an induction. Whether such an effect is observable and consequential with the other structural analogues of heme, some of which are also strong inducers of HO-1, would be important to understand in the context of selecting a safe drug with a short duration of action, no lingering side effects on other biologic processes, and no long-term alterations in HO-1 gene expression secondary to drug exposure in a critical period.
Besides a wide spectrum of HO inhibitory potential among the different Mps, for the two isozymes, the route of administration can also influence the bioavailability and efficacy of the drugs. For example, oral administration may be possible with ZnBG and with chromium mesoporphyrin (CrMP) (52,53,54), but with the current formulations, is unlikely to be possible for SnMP or ZnPP. The packaging of Mps for targeting particular tissues, such as the spleen, is also possible. Liposomes have been reported as useful for this purpose (55), and there are probably other targeting approaches that could alter distribution by the various routes of administration.
Another factor to consider when choosing a drug for hyperbilirubinemia is the fact that inhibition of HO in the liver and spleen leads to the proportional excretion of undegraded heme in bile (56). Thus, it is important to inhibit HO activity in the intestine so that heme reaching the intestine is not catabolized to bilirubin, which can be recirculated. Little is known about the effect that most of these drugs have on intestinal HO; however, it is likely that inhibition occurs sufficiently to ensure the overall attenuation of bilirubin production in the clinical setting.
The ideal chemotherapeutic agent should have a relatively short duration of action and have a limited residence time. Unfortunately, little is known about the distribution
and duration of action and metabolism of most of the Mps (57,58). Of the synthetic analogues, only cobalt protoporphyrin, besides heme, appears to be a substrate for HO and is therefore metabolized like heme. It is also likely that developmental changes may affect the pharmacokinetics of the various heme analogues, and more information is needed, in particular about the retention of SnMP after administration.
and duration of action and metabolism of most of the Mps (57,58). Of the synthetic analogues, only cobalt protoporphyrin, besides heme, appears to be a substrate for HO and is therefore metabolized like heme. It is also likely that developmental changes may affect the pharmacokinetics of the various heme analogues, and more information is needed, in particular about the retention of SnMP after administration.
In summary, the ideal compound should have a low I50 (dose for 50% inhibition of HO activity); should not be a photosensitizer; should be orally absorbable; should not cross the blood–brain barrier; should be short acting and easily excretable; should not be degraded with subsequent release of the sequestered metal iron; should not substantially upregulate HO-1, mRNA, protein, or activity; and should not affect other enzyme systems (26,41). To date, no compound seems to meet all the ideal characteristics, and compounds vary in their fulfillment of these criteria. Nonetheless, SnMP is the only compound approved as an investigational drug. It has a low I50, does not appear to cross the blood–brain barrier, but it possesses photoreactive properties, and upregulates HO-1 (26,47). ZnPP is naturally occurring but has only moderate inhibitory potency. Nevertheless, it is the least likely to be toxic and is not a photosensitizer in vivo. However, it is also unstable under acid (gastric) conditions, cannot be absorbed orally, and upregulates HO-1, but only very briefly. Its formulation is difficult and may limit its usefulness as a drug. ZnBG has very high potency, is orally absorbable, but it is a photosensitizer (50) and increases HO-1 transcription minimally (48). Thus, this compound has a promising potential, as it also contains a biocompatible, essential metal. CrMP is also an interesting compound, which may have promise. It has high inhibitory potency, is orally absorbable, does not cross the blood–brain barrier, and is photochemically inactive. It also does not upregulate HO-1 (53,54). At low doses, it affects only HO-1 and does not affect the activity of NOS or sGC, like the zinc and tin analogues and their derivatives.
D-Penicillamine
D-Penicillamine is a chelating agent in use since the 1950s in the treatment of Wilson disease and heavy metal intoxication. It has also been used to treat cystinuria and rheumatoid arthritis. In the early 1970s, D-penicillamine was described as a therapy for neonatal hyperbilirubinemia in Europe (59). Further studies provided a likely mechanism of action for its use in the treatment of hyperbilirubinemia, indicating that a 3-day course of the drug significantly reduced HO activity levels in neonatal, but not adult, rats (60). A study of D-penicillamine to reduce severe hyperbilirubinemia was conducted in 120 full-term infants with ABO hemolytic disease over a 5-year period, using 60 untreated infants from the first part of the study period as historical controls (59). If initiated within the first 24 hours of life, treatment with D-penicillamine was associated with significantly reduced PTB levels and a decreased need for exchange transfusion in this population. Although the proposed mechanism in its action in decreasing PTB levels may be through inhibition of HO activity, this finding has not been confirmed through direct measurement. Its administration (300 to 400 mg per kg per day) has been associated with amelioration of neonatal hyperbilirubinemia; however, its efficacy has not been well proven, especially in light of the considerable risks associated with its use in neonates.
Numerous cutaneous lesions, including urticaria, macular or papular reactions, pemphigoid lesions, and dermatomyositis, have been reported with long-term use of D-penicillamine (61). Furthermore, significant renal, hepatic, and hematologic complications, including nephrotic syndrome, Goodpasture syndrome, elevation of liver enzymes, aplastic anemia, and thrombocytopenia, have been associated with prolonged therapy for rheumatoid arthritis. The effect of short-term therapy on liver function has been reported in 20 term or near-term infants and on renal function in 30 term or near-term infants (62). Liver function tests were found to be unchanged after 4.3 ± 1.7 days of treatment with D-penicillamine at a dose of 300 mg per kg per day. Cholesterol, blood urea nitrogen, and creatinine levels were also unaffected after 2.8 ± 1.1 days of D-penicillamine treatment at the same dose. The in vitro effect of the drug on human peripheral granulocytes was also investigated. Superoxide anion generation and β-glucuronidase release were both found to be significantly increased at all concentrations of D-penicillamine; however, phagocytic or killing activity of the granulocytes appeared to be unaffected by the drug.
A decreased incidence of retinopathy of prematurity (ROP) was unexpectedly noted among very low-birth-weight infants treated with D-penicillamine in later studies (63,64). Recently, a meta-analysis of the effect of prophylactic D-penicillamine on the incidence of ROP in infants of less than 2,000 g birth weight was undertaken (65). This review concluded that treatment was unlikely to affect survival and may reduce the incidence of ROP among survivors. It is important to note, however, that these conclusions were based on the findings of only two randomized trials, and that no conclusion could be reached regarding the effect of D-penicillamine on severity of ROP. Further studies are required to fully evaluate the possible efficacy and potential side effects of D-penicillamine in the neonatal population before including this drug in the therapeutic armamentarium.
Other Nonmetalloporphyrin Inhibitors
Peptide inhibitors, originally developed for use in transplantation survival studies from the immunomodulatory peptide 2702.75–84, have been shown to be immunosuppressive in vitro and in vivo (66). Some of these compounds, such as D2702.75–84, can bind heat shock protein 70 and have also been found to inhibit HO activity in vitro in a dose-dependent manner. However, similar to what has been found with some Mps, administration of peptides in mice resulted in an upregulation of HO-1 mRNA and protein, as well as HO activity in liver, spleen, and kidney. Consequently, human studies using these peptides for the treatment of hyperbilirubinemia have not been performed.
Originally designed to inhibit cholesterol production (67,68,69), imidazole dioxolanes, which are structurally different from Mps, have also been found to inhibit in vitro
(70,71,72,73) and in vivo (74) HO activity. These compounds have been observed to have a high selectivity for inhibiting the inducible HO-1. Some of these compounds have been found to affect other important enzymes, such as NOS and sGC, in rat tissues (73), whereas others, such as Azalanstat, inhibit in vivo HO activity, but only at a high dose, and also can induce HO-1 gene transcription (74).
(70,71,72,73) and in vivo (74) HO activity. These compounds have been observed to have a high selectivity for inhibiting the inducible HO-1. Some of these compounds have been found to affect other important enzymes, such as NOS and sGC, in rat tissues (73), whereas others, such as Azalanstat, inhibit in vivo HO activity, but only at a high dose, and also can induce HO-1 gene transcription (74).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


