Drug Therapy in the Newborn
Robert M. Ward
Ralph A. Lugo
The developmental uniqueness of the neonate has tremendous impact on drug therapy. This uniqueness and the potential for dramatic and rapid developmental changes beginning shortly after birth defy accurate generalizations, and illustrate the need for age-specific studies in the increasingly premature patients surviving today. These developmental changes affect all aspects of drug action, from absorption and protein binding to receptor interaction and elimination. The impact of these developmental changes on drug action and the use of pharmacokinetics to adjust drug dosages will be outlined to guide the clinician between the Charybdis of ineffective therapy and the Scylla of drug overdose and toxicity (see also Appendix H).
PRINCIPLES OF PHARMACOLOGY APPLIED TO NEONATES
Free-Drug Theory and Protein Binding
Most clinical drug assays measure both bound and unbound drug; however, it is only the nonprotein-bound or free-drug molecules that are active i.e., cross membranes, bind to receptors to exert pharmacologic action, undergo metabolism and excretion (1). Serum protein binding usually is a rapidly reversible process, so that additional drug is released to replace the unbound drug removed by distribution into tissue or by elimination. The rate of release from serum protein binding usually is much faster than the rate of transfer across membranes. Seldom is the rate of release from serum proteins so slow that it limits the availability of drug molecules for transfer across membranes to exert pharmacologic effects (2).
For most drugs in premature infants, the percentage of unbound drug in the circulation is greater than in adults, because both the amount and the binding affinity of circulating proteins are decreased (3). For example, the albumin of term newborns, compared to that of adults, binds less theophylline, warfarin and sulfonamides but similar amounts of diazepam (4,5). Because the effects of a drug are related to the amount of unbound drug reaching the site of action, some drug effects in newborns may be explained only by measuring circulating concentrations of free drug. Furthermore, circulating total drug concentrations that are in the therapeutic range for adults or older children may represent free-drug concentrations that are in a toxic range in the premature neonate (5).
Absorption
In drug treatment, absorption refers to the transfer of drug from the site of administration into the circulation. The rates of drug absorption are related to several factors, beginning with the route of administration and including the same characteristics that influence transfer of any substance across lipid bilayers: degree of ionization, molecular weight, lipid solubility, concentration gradient, and active transport.
Enteral
Enteral drug treatment of neonates may not produce reliable and reproducible circulating drug concentrations for a variety of reasons. Although most studies of enteral drug therapy have been conducted in adults, many of the problems of enteral drug administration identified from these studies are likely to occur in neonates.
Intestinal villi and microvilli increase the surface area of the gastrointestinal tract, so that rates of drug absorption are usually much greater from the intestine than from the stomach. Delayed gastric emptying slows passage of drug into the intestine, which prolongs the absorption phase of many drugs. Elimination begins during this absorption phase, so that delayed gastric emptying reduces the area under the curve (AUC) for circulating drug concentration vs. time. This reduces the desired therapeutic effect for many
drugs whose effects are directly proportional to the AUC. Gastroesophageal reflux is common in neonates and may be associated with delayed gastric emptying that reduces the therapeutic effects of drugs administered orally. Few studies have addressed this aspect of drug treatment of newborns (6).
drugs whose effects are directly proportional to the AUC. Gastroesophageal reflux is common in neonates and may be associated with delayed gastric emptying that reduces the therapeutic effects of drugs administered orally. Few studies have addressed this aspect of drug treatment of newborns (6).
Additional problems, unique to the immature patient, may affect enteral drug treatment of newborns. Neonates, especially premature neonates, malabsorb fat which may alter enteral drug absorption. Elevated right atrial pressure, leading to passive congestion of hepatic and mesenteric circulations, often reduces enteral drug absorption in adults. Prolonged enteral drug administration often is necessary for treatment of infants with chronic disorders. These include bronchopulmonary dysplasia (BPD) and congestive heart failure, which may increase right atrial pressure and cause intestinal venous congestion that decreases enteral drug absorption and bioavailability. Therefore, larger doses may be required to achieve the desired therapeutic response. This phenomenon has been reported with furosemide in an infant with BPD, who required a six-fold higher enteral dose to reach plasma concentrations comparable to a 1 mg/kg dose administered intravenously (i.v.) (6).
Intramuscular
Intramuscular drug absorption is directly proportional to blood flow and the surface area of the drug deposited in the muscle (7). Although intramuscular drug administration is often considered more reliable than enteral, the sick or hypothermic neonate with limited muscle mass and poor perfusion of muscle may not absorb intramuscular doses rapidly or completely. As a result of limited amounts of muscle, injections intended for the muscle may enter subcutaneous tissue, from which absorption is slow and unpredictable. Caustic drugs (e.g., phenytoin, pH = 12) damage surrounding tissues and isolate the dose of drug from blood flow, or precipitate to a chemical form that is absorbed very slowly in what has been described as a depot effect (7). Intramuscular injection sites in neonates may leave sterile abscesses that later require surgical repair. In general, prolonged intramuscular administration of drugs in neonates should be avoided.
Intravenous
Intravenous drug administration is most likely to ensure effective drug therapy in neonates. Although this route of drug treatment is the most reliable, certain problems must be recognized that are unique to neonates. The infusion rate for i.v. fluids in extremely small neonates is so slow that drug doses injected distant from where the i.v. enters the vessel or up the i.v. tubing away from the patient, may not reach the circulation for several hours (8).
The most reliable method for administering medications intravenously to neonates is to use a small-volume syringe pump and micro-bore tubing connected as close to the patient as possible. If the syringe is prepared to contain the exact dose, the tubing must be flushed following drug administration to ensure complete drug delivery. Alternatively, it may be preferable to include overfill in the syringe so that the tubing may be primed prior to drug administration. Thus, once the drug is infused, the tubing will contain extra drug that may be discarded without the necessity of flushing.
Distribution
Distribution is the partitioning of drug from the circulation into various body fluids, organs, and tissues (9). At equilibrium, this distribution is related to organ blood flow; pH and composition of body fluids and tissues; physical and chemical properties of the drug including lipid solubility, polarity, and size; and the extent of binding to plasma and tissue proteins.
Dramatic developmental changes in body composition of newborns influence the distribution of polar and nonpolar drugs within the body. At 24 weeks of gestation, water comprises about 89% of body weight, with 0.1% to 0.5% as fat (10,11). Thus, water-soluble drugs that distribute primarily into extracellular fluid will have larger distribution volumes in premature neonates. By 40 weeks of gestation, the body is approximately 75% water and 15% fat, compared to adults in whom the body is about 65% water and the fat content is variable. The low fat content of the brain of the extremely premature newborn may affect the distribution and effects of centrally active drugs, such as barbiturates and gaseous anesthetics (12).
Metabolism
Many drugs require biotransformation to more polar forms before they can be eliminated from the body. Biotransformation reactions are designated phase I reactions that make the drug more polar through oxidation, reduction, or hydrolysis; or phase II, conjugation reactions, such as glucuronidation, sulfation, and acetylation (9). The liver is the primary site for biotransformation, however other organs are also involved. As early as 9 to 22 weeks gestation, metabolic enzyme activities of the fetal liver vary from 2% to 36% of adult activity (13). This variation precludes broad generalizations regarding hepatic drug metabolism in premature newborns. Over the past decade, intense research into the biochemistry of drug metabolism has revealed multiple forms of cytochrome P450 with different substrate specificities and different activities during development from the fetus to the adult (14,15,16,17). Clinicians should have an understanding of P450 nomenclature in addition to understanding which isoforms are responsible for metabolism of commonly used drugs. Both induction and inhibition of specific isoforms may require more frequent therapeutic drug monitoring and dosage adjustments.
Cytochromes P450
Quantitatively, the most important of the phase I enzymes are the cytochromes P450, a superfamily of heme-containing proteins that catalyze the metabolism of many lipophilic substances. The cytochrome P450 isozymes are designated as CYPs that are grouped by the degree of identity in their amino acid sequences. CYP is followed in order by (a) an Arabic number representing the gene family for enzymes with greater than 40% identity; (b) a letter which indicates the subfamily for enzymes with greater than 55% identity; and (c) sequential numbering of the P450 enzymes for different isoforms within each subfamily (9,16). Isozymes which are important in human drug metabolism are found mostly in the CYP1, CYP2, and CYP3 gene families. Table 56-1 outlines the P450 isozymes and their common substrates in the newborn.
Research in the early 1970s revealed that newborn infants have significantly reduced total quantities of cytochrome P450 in liver microsomes (18). This hemoprotein increases with gestational age but reaches only 50% of adult values at term (18). Reduced cytochrome P450 in neonates explains the low clearance and significantly prolonged half-lives of theophylline, caffeine, phenytoin, phenobarbital and other substances that are metabolized via cytochrome P450 (5,19,20,21). Although newborns are poor metabolizers of many xenobiotics, specific P450 cytochromes exhibit unique developmental patterns during gestation and postnatal life that invalidate broad generalizations about drug metabolism. Table 56-1 outlines important developmental patterns for many of these enzymes.
TABLE 56-1 DEVELOPMENTAL PATTERNS FOR IMPORTANT CYTOCHROME P450 ENZYMES IN THE NEONATE | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||
Ontogeny of Important P450 Cytochromes
Cytochrome P4501A2 is extensively involved in the metabolism of caffeine (1,3,7-trimethylxanthine) (22,23) and theophylline (1,3-dimethylxanthine) (24,25), drugs that are commonly used to treat neonatal apnea and bradycardia (see methylxanthines). CYP1A2 is not significantly expressed in human fetal liver and expression is very low in neonates reaching only 50% of adult activity by 1 year of age (25,26). Metabolically, this limits N-3- and N-7-demethylation of caffeine in the newborn period (23). Caffeine elimination in both preterm and term infants is significantly prolonged (27). Maturation of this pathway to adult levels occurs between four and six months postnatally (28,29). A similar pharmacokinetic trend is noted with theophylline in which 3-demethylation and 8-hydroxylation are catalyzed by CYP1A2 (24,25). Clinically, theophylline clearance and urine metabolite patterns reach adult values by 55 weeks postconceptional age or approximately 4 to 5 months postnatally (30).
Other P450 enzymes which appear to be reduced or absent in the fetus include CYP2D6 and CYP2C9 (14,15,31). The former is responsible for the metabolism of numerous important therapeutic compounds including =β-blockers, antiarrhythmics, antidepressants, antipsychotics, and codeine. Although CYP2D6 is absent in the fetal liver and appears to be expressed postnatally (15), activity remains low for an extended period (see Table 56-1) (32). In contrast to the slow development of CYP1A2 and CYP2D6, other enzymes such as CYP2C9 (responsible for the metabolism of nonsteroidal antiinflammatory drugs,
warfarin, and phenytoin) develop more rapidly after birth. For example, although CYP2C9 is not significantly present during fetal life (31), phenytoin pharmacokinetic data in newborns suggest rapid enzyme maturation within the first weeks of life (20,33).
warfarin, and phenytoin) develop more rapidly after birth. For example, although CYP2C9 is not significantly present during fetal life (31), phenytoin pharmacokinetic data in newborns suggest rapid enzyme maturation within the first weeks of life (20,33).
For drug metabolism, the most important of the cytochromes P450 is CYP3A, because of the large number of therapeutic substrates for this subfamily of enzymes (see Table 56-1). Additionally, CYP3A accounts for the majority of P450 cytochromes present in the adult human liver (9). Unlike most of the other important cytochromes, CYP3A is functionally present during embryogenesis, primarily as CYP3A7 (34). CYP3A activity is detectable in large amounts as early as 17 weeks gestation, and reaches 75% of adult activity at 30 weeks gestation (15). In vivo, CYP3A activity appears to be mature at birth (17,35). Postnatally, there is a poorly understood transition from the fetal CYP3A7 to the predominant adult isoform CYP3A4.
Phase II Reactions
The phase II reactions are known as synthetic or conjugation reactions and function to increase the hydrophilicity of drug molecules which facilitates renal elimination (9). The phase II enzymes include glucuronosyltransferase, sulfotransferase, N-acetyltransferase, glutathione S-transferase, and methyl transferase. Although the ontogeny of phase II reactions as a group is not well studied, developmental changes during infancy influence drug clearance (see Table 56-2).
Most conjugation reactions show low activity during fetal development, although sulfation is relatively active in the fetus (16,36,37). One of the most common synthetic reactions involves conjugation with uridine diphosphoglucuronosyltransferases (UDP-GT). This enzyme system which is comprised of numerous isoforms is also responsible for glucuronidation of endogenous compounds such as bilirubin (38). Although UDP-GT activity for bilirubin develops relatively rapidly after birth (36), the ability of the infant to glucuronidate xenobiotics is significantly limited during the newborn period. Thus, without dosage adjustments, drugs may accumulate to toxic concentrations during the newborn period. A tragic example occurred in the early 1960s when newborns received standard pediatric doses of chloramphenicol and developed fatal circulatory collapse, a condition known as the gray baby syndrome (39,40,41). The clearance of chloramphenicol is low during the neonatal period and dosage adjustments are necessary in preterm and full-term infants to avoid chloramphenicol toxicity (42).
TABLE 56-2 DEVELOPMENTAL PATTERNS FOR IMPORTANT CONJUGATION REACTIONS IN THE NEONATE | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Other drugs used in the newborn period that undergo glucuronidation include morphine, acetaminophen, and lorazepam. The major metabolic pathway of morphine in children and adults is glucuronidation in the 3- and 6-position (43,44). However, neonates have limited ability to glucuronidate morphine and thus require dosage adjustment (45,46,47). Morphine clearance (45,48), in particular 3- and 6-glucuronide formation, are depressed at birth and increase with birth weight (47), gestational age (49), and postnatal age (43,46). Morphine’s clearance and half-life begin to approach adult values after the age of 1 month (46,50), although other reports indicate that adult values are not reached until at least 5 to 6 months (45,51). Overall, the maturation of glucuronosyltransferase enzymes is isoform-specific; however, adult activity is usually achieved by 6 to 18 months of age (16).
In contrast to glucuronosyltransferase, the sulfotransferase enzyme system is well developed in the newborn and may compensate for limited glucuronidation, as is the case with the metabolism of acetaminophen. Although acetaminophen is primarily glucuronidated in adults, the half-life of acetaminophen is only moderately prolonged in term newborns as compared to older infants and adults (52,53,54). In the neonate, this is explained by a relatively large formation rate constant for the acetaminophen-sulfate leading to a greater percent of the dose excreted as the acetaminophen-sulfate conjugate (52,53). Preferential sulfation of acetaminophen continues into childhood (53,55).
Alterations in Biotransformation
Biotransformation reactions, especially those involving certain forms of cytochrome P450, are often inducible before birth through maternal exposure to drugs, cigarette smoke, or other xenobiotic inducing agents. Biotransformation reactions may also be induced by postnatal drug exposure (see Tables 56-1 and 56-2) and may be slowed postnatally by hypoxia/asphyxia, organ damage, and/or critical illness. Additional postnatal changes in hepatic blood flow, protein binding, and/or biliary function may also significantly alter drug elimination. Additional studies in premature neonates are required to generate the population pharmacokinetic data necessary to design safe and effective pharmacotherapeutic regimens.
Excretion
Excretion involves elimination of drug from the body by several potential routes, including the biliary tract, lungs, and kidneys. Both unchanged and metabolized forms of drug may be excreted, but only unbound drug undergoes filtration and tubular transport. Glomerular and tubular function are decreased at birth, both in absolute terms and after normalization to body mass (56,57). Glomerular filtration in newborns averages 30% of the adult rate after normalization to body surface area. Birth accelerates maturation of glomerular filtration through an increase in cardiac output, decreased renal vascular resistance, redistribution of intrarenal blood flow, and changes in the intrinsic function of the glomerular basement membrane (56). Renal tubular maturation seems to proceed more slowly than glomerular maturation after birth (56). This produces an imbalance in glomerular and tubular function that persists for several months. Because most low molecular weight unbound molecules are filtered, tubular reabsorption exerts a profound influence on the elimination rate for many drugs. Additionally, hypoxemia, nephrotoxic drugs, and underperfusion may alter renal function of newborns, which prevents accurate prediction of the rates of drug elimination after birth.
PHARMACOKINETICS
Pharmacokinetics describes the changes in drug concentrations within the body with time. These concepts are presented as a general overview to assist the clinician with dose adjustments and practical interpretation of therapeutic drug monitoring (58,59,60). The more rigorous mathematical intricacies of pharmacokinetics are covered elsewhere (61,62,63,64). Although a drug may penetrate several body fluids and tissues at different rates, the change in its circulating concentration is used to characterize its kinetics and to guide dosages. The rate of removal of drug from the circulation usually fits either first-order or zero-order exponential mathematical equations. These two types of equations describe two different processes that have important implications for dosage regimes.
Rates and Distribution
First-Order Kinetics
Most drugs are cleared from the body with first-order exponential rates. Exponential clearance indicates that a constant fraction or constant proportion of drug is removed per unit of time. This means that the higher the concentration, the greater the amount of drug removed from the body. Such changes in concentration fit exponential equations of the following form:
where Ct is the concentration at a particular time t, C0 is the starting concentration, which is a constant, and k is the elimination rate constant with units of 1/time. First-order indicates that the exponent is raised to the first power (-kt in Eq. 1). First-order exponential equations, such as Eq. 1, may be solved by taking the natural logarithm of both sides.
This transforms the equation to that of a straight line (y = mx+b). If ln (i.e., natural logarithm) C is graphed vs. time, the slope is -k, and the intercept is ln C0. If log i.e., common logarithm C is graphed vs. time, the slope is -k/2.303, because ln x equals 2.303 log x. When graphed on linear-linear axes, exponential rates are curvilinear and on semilogarithmic axes, they produce a straight line.
Half-Life
One of the more familiar exponential rates used clinically is the half-life, i.e., the time for a drug concentration to decrease by one-half. Half-life is a first-order kinetic process, because the same proportion or fraction of the drug is removed during equal time periods. At higher concentrations, a greater amount is removed during a single half-life than when the concentration is lower. For example a drug concentration may decrease by 200 from 400 to 200 in one half-life, and decrease by 100 from 200 to 100 in the next half-life (Fig. 56-1).
Half-life can be determined by several methods. If concentration is converted to the natural logarithm of concentration and graphed vs. time, as described in Eq. 2, the slope of this graph is the elimination rate constant, k. Usually at least three concentration-time points are needed to determine the slope accurately; however, in clinical practice, k is often determined from just two concentrations obtained during the terminal elimination phase. To increase the accuracy of the latter, at least one half-life should elapse between concentration-time points. With multiple data points, the slope of ln C vs. time may be calculated easily by least-squares linear regression analysis. Half-life (t1/2) may be calculated from the elimination rate constant, k (1/time), as follows:
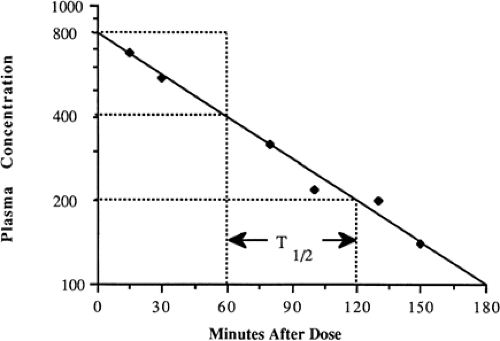 Figure 56-1 Graph method for estimation of half-life. From Ward RM. Pharmacologic principles and practicalities. In: Taeusch HW, Ballard RA, Avery ME, eds. Shaffer and Avery’s diseases of the newborn. 6th ed. Philadelphia: WB Saunders, 1991:289, with permission. |
Half-life may be determined graphically from a series of drug concentrations graphed on semilogarithmic axes. With multiple data points, the best-fit line is determined either visually or by linear regression analysis. The times corresponding to carefully chosen concentrations are then used to estimate the interval required for the concentration to decrease by one-half. In Fig. 56-1, this is illustrated by the times corresponding to concentrations of 400, 200, and 100, estimated by the horizontal broken line intercepts with the concentration line and the intercepts with the time axis indicated by the vertical broken lines. Note that the concentrations decrease by 50% every 60 minutes, so that t1/2 equals 60 minutes.
First-Order Single-Compartment Kinetics
The number of compartments refers to the number of exponential equations required to describe the observed changes in concentration. These compartments theoretically represent a group of similar tissues, fluids, and/or organs and may be correlated with different anatomic fluids and tissues. Although multiple transfers of drug among tissues and body fluids may be occurring, a drug’s clearance may fit first-order, single-compartment kinetics if it distributes rapidly and homogeneously within the circulation from which it is removed through metabolism or excretion. This may be judged visually, if a semilogarithmic graph of a series of concentrations fits a single straight line. Kinetics may falsely appear to be single-compartment if drug concentrations are not measured quickly enough after i.v. administration to detect the initial distribution phase.
First-Order Multicompartment Kinetics
If drug clearance from the circulation is studied carefully, with measurement of concentration several times within the first 15 to 30 minutes after i.v. administration and during the next several hours, two or more rates of clearance often are detected by a change in slope of a semilogarithmic graph of concentration vs. time (Fig. 56-2). The number and nature of the compartments for the clearance of a drug do not necessarily correspond to specific body fluids or tissues. When two first-order exponential equations are required to describe the clearance of drug from the circulation, the kinetics are designated first-order and two-compartment i.e., central and peripheral compartments and are represented by the following equation (58):
In Eq. 4, C is concentration, t is time after the dose, A is the concentration at time 0 for the distribution rate represented by the broken line graph with the steepest slope, is the rate constant for distribution, B is the concentration at time 0 for the terminal elimination rate, and β is the rate constant for terminal elimination. Rate constants indicate the rate of change in concentration and correspond to the slope of the line divided by 2.303 for logarithm concentration vs. time.
Such biphasic kinetics are observed commonly for drugs that rapidly distribute out of the central compartment (blood volume + extracellular fluid of highly perfused organs) after i.v. administration (58). For such drugs, the initial rapid decrease in concentration is referred to as the alpha distribution phase and represents primarily distribution to the peripheral (tissue) compartments in addition to drug elimination. After the inflection point in the slope and during the terminal (beta) phase of the curve, elimination accounts for most of the change in drug concentration. The alpha distribution rate constant (α) (see Fig. 56-2) can be determined from the slope of the line generated by subtracting concentrations from the beta elimination phase from those during the alpha distribution phase. A more detailed mathematical discussion may be found elsewhere (58,61).
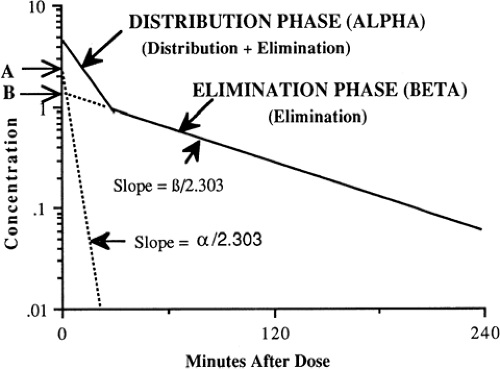 Figure 56-2 Multicompartment first-order exponential kinetics. From Ward RM. Pharmacologic principles and practicalities. In: Taeusch HW, Ballard RA, Avery ME, eds. Schaffer and Avery’s diseases of the newborn. 6th ed. Philadelphia: WB Saunders, 1991:289, with permission. |
Although many drugs demonstrate multicompartment kinetics, the intensive blood sampling required to fit data to more than one compartment is not clinically feasible, particularly in newborns. Furthermore, the mathematical complexity of two compartment models makes this kinetic approach clinically impractical. To minimize cost and simplify pharmacokinetic calculations, only two plasma concentrations (peak and trough) are usually obtained for therapeutic monitoring of commonly used drugs, e.g., gentamicin and vancomycin. Accordingly, a one-compartment model is assumed and the elimination rate constant (k) is determined from the slope of these points plotted on a semi-logarithmic scale. Because the elimination rate constant should be determined from the terminal elimination phase, it is important that peak concentrations of multicompartment drugs not be drawn prematurely, i.e., during the initial distribution phase. If drawn too early, the concentrations will be higher than those during the terminal elimination phase (see Fig. 56-2), which will overestimate the slope and the terminal elimination rate constant. Clinically, this is not usually problematic with gentamicin because the initial distribution phase commonly occurs during the 30- to 60-minute infusion (65). Thus, a peak concentration obtained 30 to 60 minutes after the end of infusion usually reflects the terminal phase of elimination. However, with vancomycin, the half-life of the initial distribution phase is approximately 0.5 hours (66). Thus, a peak vancomycin concentration drawn prematurely may lead to error in estimating the pharmacokinetic parameters.
Zero-Order Kinetics
Some drugs are eliminated by a constant amount per unit time, rather than a constant fraction. Such rates are zero-order and the following equation can be used to calculate the change in the amount of drug in the body (61):
in which dA is the change in the amount of drug in the body (mg), dt is the change in time, and k0 is the elimination rate constant with units of amount/time. After solving this equation, it has the following form:
in which A0 is the initial amount in body and A is the amount of drug in the body (mg) at time t.
Zero-order kinetics also is referred to as saturation kinetics, because it may occur when excess amounts of drug saturate the capacity of metabolic enzymes or transport systems so that only a constant amount of drug is metabolized or transported per unit of time. This may be detected graphically from a serum concentration vs. time plot in which zero-order elimination is linear on linear-linear axes and is curved when graphed on logarithmic-linear (i.e., semilogarithmic) axes. Clinically, zero-order elimination may be observed after administration of exces sive doses, or during dysfunction of the organ of elimination without a decrease in dosage. Certain drugs administered to newborns exhibit zero-order kinetics at therapeutic doses, and may accumulate to excessive concentrations (Table 56-3). Some drugs, e.g., phenytoin, may exhibit Michaelis-Menten kinetics, i.e., first-order at low concentrations and zero-order after enzymes are saturated at higher concentrations. For these drugs, a small increment in dose may cause disproportionately large increments in serum concentrations (Fig. 56-3).
TABLE 56-3 DRUGS THAT DEMONSTRATE ZERO-ORDER (SATURATION) KINETICS WITH THERAPEUTIC DOSES IN NEWBORNS | ||
|---|---|---|
|
Apparent Volume of Distribution
Volume of distribution does not necessarily correspond to a physiologic body fluid or tissue volume, hence the designation “apparent.” The apparent volume of distribution (Vd) is a mathematical term that relates the dose to the circulating concentration observed immediately after intravenous administration. It might be viewed as the volume of dilution. For drugs such as digoxin, Vd in neonates may reach 10 L/kg, a physical impossibility. Such a large Vd occurs when drug concentrates outside of the plasma compartment, for example bound to red blood cells or in tissue. This emphasizes the mathematical nature of Vd. The units used to express concentration are amount/volume and may help to remind the reader of the following equation that expresses the relation between dose in amount/kg and the Vd in volume/kg that dilutes the dose to produce the concentration:
To facilitate canceling units, concentration is expressed with the unconventional units of milligrams per liter rather than micrograms per milliliter, because they are equivalent. This equation serves as the basis for most of the pharmacokinetic calculations, because it is easily rearranged to solve for Vd and Dose. It is also important to note that this equation represents the change in concentration following a rapidly administered intravenous dose. Following an intravenous infusion, e.g., vancomycin or gentamicin, a more complex exponential equation may be required to account for drug elimination during the time of infusion (61).
However, such equations are needed only when the drug is rapidly eliminated, i.e., the duration of infusion approaches the drug’s half-life. In neonates, who have relatively slow drug elimination, only a small fraction of drug is eliminated during the time of infusion, and such adjustments can be omitted. Accordingly, we may use the simpler and more practical equation to estimate pharmacokinetic parameters.
However, such equations are needed only when the drug is rapidly eliminated, i.e., the duration of infusion approaches the drug’s half-life. In neonates, who have relatively slow drug elimination, only a small fraction of drug is eliminated during the time of infusion, and such adjustments can be omitted. Accordingly, we may use the simpler and more practical equation to estimate pharmacokinetic parameters.
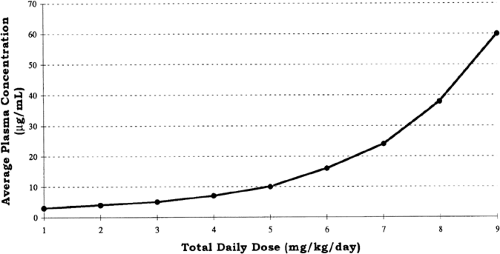 Figure 56-3 Nonlinear (Michaelis- Menten) kinetics illustrated for a hypothetical drug. As dosage increases above 3 mg/kg/day, there is a disproportionate increase in average plasma concentration as elimination changes from a first-order to a zero-order process. |
Knowledge of the apparent distribution volume is essential for dosage adjustments. Vd may be calculated by rearranging Eq. 7.
The concentration after drug infusion, C (postdose), must be measured after the distribution phase to avoid overestimating the peak concentration that would result in an erroneously low Vd. For the first dose, the predose concentration is zero.
Pharmacokinetic Example
To illustrate the practical application of the principles outlined above, we recommend a simple four-step approach: (a) calculate Vd; (b) calculate half-life; (c) calculate a new dose and dosing interval based on a desired peak and trough; (d) check the peak and trough of the new dosage regimen.
For example, gentamicin 2.5 mg/kg every 12 hours was administered i.v. over 30 minutes. The following plasma concentrations were measured on the third day of treatment (presumed steady-state). The predose or trough concentration was 1.8 mg/L; the peak concentration, measured 30 minutes after the end of the infusion, was 4.8 mg/L.
Step 1: Substituting the data into Eq. 8, we calculate Vd.
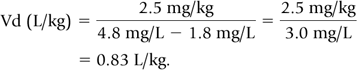
Step 2: At steady-state, trough concentrations remain unchanged from one dose to the next. Therefore, for the purpose of these calculations, steady-state trough concentration may be understood to follow the peak concentration. Thus, the time between the peak and trough concentrations is 11 hours, i.e., 12 hours minus 0.5 hour infusion, minus 0.5 hour to peak concentration. The plasma concentration decreased from 4.8 to 2.4 mg/L in one half-life, and then from 2.4 to 1.2 mg/L in a second half-life. The trough of 1.8 was reached approximately half way between the first and second half-lives. Because 1.5 half-lives elapsed during the 11 hours between the peak and trough, the half-life is approximately 11 hours/1.5 half-lives, or 7.3 hours.
Step 3: A new dosage regimen must be calculated if the concentrations are unsatisfactory. Accordingly, one must decide on a desired peak and trough concentration. If, for example, the desired gentamicin peak and trough concentrations were 6.5 mg/L (5-10 mg/L) and 1.5 mg/L (1-2 mg/L), respectively, then Eq. 8 may be rearranged to solve for the new dose.
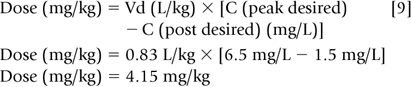
Increasing the dose to 4.15 mg/kg (66% increase) will lead to a higher trough if the dosage interval is kept at 12 hours. Because trough concentrations above 2 mg/L are associated with ototoxicity and nephrotoxicity, the dosage interval needs to be increased. Aminoglycosides, in general, are dosed every 2 to 2.5 half-lives for neonates. However, to reduce the risk of administration errors, one should choose a conventional dosage interval that approximates 2 to 2.5 half-lives, i.e., every 8, 12, 18, 24, or 36 hours. In the above example, dosing every two half-lives would require a 15-hour dosage interval that might lead to administration errors. Thus, the dosage interval should be increased to 18 hr, an interval that corresponds to approximately 2.5 half-lives.
Step 4: Mathematically estimating peak and trough concentrations with the new regimen provides a good double check against a math error. Because 18 hours was
chosen as the new interval, waiting 18 hours after the previous dose to begin the new regimen is reasonable. At that time, approximately 2.5 half-lives after the measured peak of 4.8 mg/L, we would expect the trough concentration to be approximately 0.9 mg/L (half-life #1: 4.8 mg/L →2.4 mg/L; half-life #2: 2.4 mg/L →1.2 mg/L, half-life #3: 1.2 mg/L →0.6 mg/L, with half of this third half-life occurring at 0.9 mg/L). Using Eq. 7, we calculate that the plasma concentration will increase by 5 mg/L after each 4.15 mg/kg dose. Accordingly, the measured peak concentration will be 5.9 mg/L after the first dose and will decrease to 1.1 mg/L over the next 2.5 half-lives (5.9 mg/L → 2.95 mg/L →1.48 mg/L →0.74 mg/L in 3 half-lives, or 1.1 mg/L in 2.5 half-lives). After the second dose of 4.15 mg/kg, the measured peak concentration will be 6.1 mg/L and will decrease to 1.15 mg/L in 2.5 half-lives. Because the trough has not changed from the previous trough, one can conclude that this dosage schedule will meet the desired therapeutic levels. Estimation of concentrations after a fraction of a half-life assumes a linear rather than logarithmic decline in concentrations and is not mathematically correct for first-order, exponential rates in which the amount of drug eliminated decreases as the concentration decreases. However, this approach is suitable for clinical applications, in which only a reasonable estimate of plasma concentration is needed for dosage adjustments.
chosen as the new interval, waiting 18 hours after the previous dose to begin the new regimen is reasonable. At that time, approximately 2.5 half-lives after the measured peak of 4.8 mg/L, we would expect the trough concentration to be approximately 0.9 mg/L (half-life #1: 4.8 mg/L →2.4 mg/L; half-life #2: 2.4 mg/L →1.2 mg/L, half-life #3: 1.2 mg/L →0.6 mg/L, with half of this third half-life occurring at 0.9 mg/L). Using Eq. 7, we calculate that the plasma concentration will increase by 5 mg/L after each 4.15 mg/kg dose. Accordingly, the measured peak concentration will be 5.9 mg/L after the first dose and will decrease to 1.1 mg/L over the next 2.5 half-lives (5.9 mg/L → 2.95 mg/L →1.48 mg/L →0.74 mg/L in 3 half-lives, or 1.1 mg/L in 2.5 half-lives). After the second dose of 4.15 mg/kg, the measured peak concentration will be 6.1 mg/L and will decrease to 1.15 mg/L in 2.5 half-lives. Because the trough has not changed from the previous trough, one can conclude that this dosage schedule will meet the desired therapeutic levels. Estimation of concentrations after a fraction of a half-life assumes a linear rather than logarithmic decline in concentrations and is not mathematically correct for first-order, exponential rates in which the amount of drug eliminated decreases as the concentration decreases. However, this approach is suitable for clinical applications, in which only a reasonable estimate of plasma concentration is needed for dosage adjustments.
Therapeutic Drug Monitoring
Circulating concentrations of drugs should be measured primarily to ensure that the treatment regime achieves concentrations that are effective in clinical situations in which drug treatment is critical, response is not immediately apparent (e.g., for culture-proven sepsis), and a good correlation exists between circulating drug concentration and desired effect. Drug concentrations should be measured to avoid toxicity when they clearly correlate with toxicity in newborns (e.g., for chloramphenicol), to verify toxicity when symptoms correspond to a known drug toxicity, or to investigate symptoms that are unexplained by the disease process. Extrapolation of toxic and therapeutic ranges from adults to neonates has led to some recommendations about therapeutic drug monitoring in newborns (e.g., for gentamicin) that are not well supported by subsequent experience (67).
Several basic requirements must be met to justify therapeutic drug monitoring in newborns and to modify drug treatment accurately based on measured circulating drug concentrations (68).
Drug analysis using small blood volumes must be accurate.
Circulating drug concentrations must correlate with both effective and toxic pharmacologic effects. This implies that the circulating total (i.e., free plus protein-bound) drug concentration correlates with the free-drug concentration at the site of drug action, such as the drug receptor or tissue site.
The therapeutic index, the concentration range between efficacy and toxicity, should be narrow.
Clinical studies should have established a concentration range for efficacy and toxicity in the population being monitored.
Pharmacokinetics are variable and unpredictable in newborns.
In the extremely premature newborn, decreased protein binding may significantly affect therapeutic drug monitoring. As a result of unpredictable decreases in protein binding associated with organ dysfunction or immaturity, free-drug concentrations may be much higher than would be predicted from the total drug concentration that usually is measured clinically. Higher percentages of free drug in the newborn may account for signs of toxicity or adequate therapeutic response at paradoxically low total drug concentrations. When available and when therapeutic concentration ranges have been established, as for phenytoin, measurement of free-drug concentrations may be helpful in newborns demonstrating signs of drug toxicity with therapeutic or subtherapeutic circulating total drug concentrations.
Effective drug therapy is measured by response, not by achieving a particular circulating drug concentration. Concentration ranges described as therapeutic are statistical ranges for drug levels that usually are effective and nontoxic. Individual patients may require drug concentrations outside these ranges to achieve optimal drug treatment.
Repetitive Dosing and Drug Accumulation
During most courses of repetitive drug therapy, the doses are administered before complete elimination of the previous one. This leads to accumulation of drug, with increasing peak and trough concentrations until a steady state concentration is reached (Fig. 56-4). The average Css can be calculated as follows (59):
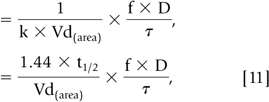
In Eq. 10 and 11, f is the fraction of the dose that is absorbed, D is the dose,τ is the dosing interval in the same units of time as the elimination half-life, k is the elimination rate constant, and 1.44 equals 1/0.693 (see Eq. 56-3). The magnitude of the average Css is directly proportional to a ratio ofτ/half-life and D. (59)
Steady State
Steady-state occurs when the amount of drug removed from the body between doses equals the amount of the dose
(64,69). Five half-lives are usually required for drug elimination and distribution among tissue and fluid compartments to reach an equilibrium. When all tissues are at equilibrium, i.e., steady-state, the peak and trough concentrations are the same after each dose. However, prior to this time, constant peak and trough concentrations after intermittent doses, or constant concentrations during drug infusions, do not prove that a steady-state has been achieved because drug may still be entering and leaving deep tissue compartments. During continuous infusion, the fraction of steady state concentration that has been reached can be calculated in terms of multiples of the drug’s half-life (59). After 3 half-lives, the concentration is 88% of that at steady state. The effect of dosage changes on drug concentrations during chronic treatment usually should not be rechecked until several half-lives have elapsed, unless elimination is impaired or toxic symptoms occur. Drug concentrations may not need to be checked if symptoms improve.
(64,69). Five half-lives are usually required for drug elimination and distribution among tissue and fluid compartments to reach an equilibrium. When all tissues are at equilibrium, i.e., steady-state, the peak and trough concentrations are the same after each dose. However, prior to this time, constant peak and trough concentrations after intermittent doses, or constant concentrations during drug infusions, do not prove that a steady-state has been achieved because drug may still be entering and leaving deep tissue compartments. During continuous infusion, the fraction of steady state concentration that has been reached can be calculated in terms of multiples of the drug’s half-life (59). After 3 half-lives, the concentration is 88% of that at steady state. The effect of dosage changes on drug concentrations during chronic treatment usually should not be rechecked until several half-lives have elapsed, unless elimination is impaired or toxic symptoms occur. Drug concentrations may not need to be checked if symptoms improve.
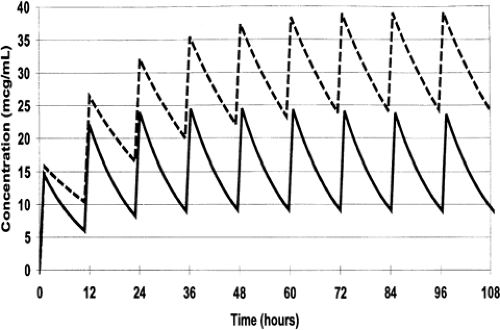 Figure 56-4 The effect of 2 different half-lives on the steady state concentrations of vancomycin during repetitive dosing with the same dose (15 mg/kg and a Vd of 0.9 L/kg. The solid line represents a half-life of 8 hours and the broken line represents a half-life of 16 hours. Peak and trough concentrations increase until steady-state is achieved, but steady-state is achieved by the fourth dose (36 hrs) with the 8 hour half-life, but not until the 7th dose (72 hours) with the 16 hour half-life |
Loading Dose
If the time to reach a constant concentration by continuous or intermittent dosing is too long, a loading dose may be used to reach a higher constant concentration more quickly. This frequently is applied to initial treatment with digoxin, which has a 35- to 69-hour half-life in term neonates and an even longer half-life in preterm newborns (70). Use of a loading dose produces a higher circulating drug concentration earlier in the therapeutic course, but the equilibration to reach a true steady-state still requires treatment for five or more half-lives. Loading doses must be used cautiously, because they increase the likelihood of drug toxicity, as has been observed with digoxin digitalizing doses (70).
Special Considerations for Neonates
Intravenous Administration
Intravenous administration of drugs is considered the most reliable route. In neonates, especially those who weigh 500 to 1000 g and receive small volumes of i.v. fluids, i.v. drug administration may not reliably deliver the dose into the circulation (8,69). Because some types of i.v. tubing may contain 12- to 15-mL of fluid and the i.v. infusion rates for 500- to 1,000-g neonates may be 1.5 to 2.5 mL/hour, infusion of a drug into the i.v. tubing several cm from the patient will markedly delay drug administration. This may confound therapeutic drug monitoring by causing the peak concentration to be less than the trough, because the dose has not reached the patient. For this reason, microbore tubing, which has a volume of 0.5 mL should be used whenever possible and should be connected to a port closest to the patient. Slow delivery of drug to the circulation may prevent the attainment of an adequate peak concentration to enhance drug diffusion into tissue down a concentration gradient. Lastly, filters present potential obstacle to effective i.v. drug treatment. Drugs may adsorb to the filter or settle to the bottom of a reservoir in the filter, out of the main flow of the infusing solution (71).
Exchange Transfusion
Because few studies have evaluated the amount of drug actually removed from neonates during an exchange transfusion, theoretical estimates have been developed (69,72). The amounts removed vary with the individual drug’s volume of distribution, rate of distribution, and circulating concentration at the time of the exchange, and the volume of blood exchanged and the rate of the exchange.
CLINICAL TOXICOLOGY
Newborn infants, both full-term and premature, present an alarmingly wide spectrum of susceptibilities to unanticipated, adverse effects from exposure to exogenous chemicals. New drugs, together with unrecognized chemicals ranging from tape remover to plasticizers, are introduced into the care of neonates each year. Prescription drug exposure of the extremely preterm neonate is extensive. In the late 1970s, neonates admitted to a neonatal intensive care unit (NICU) received 1 to 26 drugs, averaging 6.2 drugs per infant (73). This drug exposure was not innocuous, because 30% of infants in the NICU manifested an adverse drug reaction, of which 15% were considered fatal or life-threatening (74). Similar drug exposure of newborns was documented in Boston from 1974 to 1977, in which infants in the intensive care nursery received more drugs (10.4/patient) than any other hospitalized children (75).
Several factors increase the susceptibility of newborns and preterm newborns to chemical toxicities. Immaturity of liver and renal function frequently delays drug elimination, which prolongs the exposure of the newborn to a drug and predisposes to drug accumulation during repeated administration. New therapeutic agents often are introduced into treatment of critically ill neonates when all other therapy has failed, although pharmacokinetic data to guide dose and dosing intervals are lacking. Without the guide of pharmacokinetic or concentration-response studies, treatment failure may be considered an indication to increase the dose, rather than the result of administering excess amounts of the drug (76).
Specific enzyme immaturities of the newborn also may place them at risk. Inadequate function of the glucuronosyltransferase system predisposes the newborn to inadequate elimination of chemicals requiring glucuronide conjugation, such as bilirubin. Circulating albumin binds bilirubin at the levels encountered in physiologic jaundice and protects the newborn from bilirubin encephalopathy as long as the blood-brain barrier remains intact (77,78). Failure to recognize the competition of drugs for bilirubin’s albumin-binding sites has led to displacement of bound bilirubin and kernicterus (79,80,81). Large doses of chloramphenicol were associated with unexplained cardiovascular collapse in infants in 1959 (40). That same year, a randomized, controlled study of antibiotics for neonatal sepsis revealed that the groups treated with chloramphenicol alone or in combination had a 60% mortality, threefold higher than neonates receiving no antibiotics (41). The no-treatment control group, however, showed the same survival as the alternate antibiotic treatment group. Inadequate glucuronide conjugation of chloramphenicol and decreased tubular secretion of conjugated chloramphenicol combined to reduce chloramphenicol elimination and led to accumulation to toxic concentrations (39). Acute intoxication with high serum chloramphenicol concentrations manifested with jaundice, vomiting, anorexia, respiratory distress, abdominal distention, cyanosis, green stools, lethargy, and ashen color (41).
The preservative, benzyl alcohol, has been implicated in a fatal syndrome in premature infants of cardiovascular collapse and death associated with metabolic acidosis, gasping respirations, thrombocytopenia, hepatic and renal failure, and progressive central nervous system (CNS) depression (82). The minimal intake to produce toxicity was estimated at 130 mg/kg/day (82). Removal of benzyl alcohol as a preservative in the fluid frequently used to flush i.v. catheters in neonates essentially has eliminated this problem. Although many nurseries and pharmacies have excluded all solutions and medications containing benzyl alcohol, the major problem seemed to lie with i.v. solutions and flush solutions. Considering the dose estimated for toxicity, exposure to the small amounts in medications (which can be estimated from the benzyl alcohol concentration and dosing volume) might pose an acceptable risk compared to the benefit of the drug treatment. Propylene glycol (PG), a solvent frequently used with water-insoluble drugs, has also been associated with toxicity in neonates (83,84). At high doses, propylene glycol is partly metabolized to lactic acid and may cause serum hyperosmolality with a marked osmolar gap, lactic acidosis, seizures, and cardiac arrhythmias (83,84,85,86). Because up to 45% of PG is eliminated renally, patients with poor renal function (including preterm neonates) may be predisposed to PG toxicity. Lorazepam, which is commonly used in neonates, contains propylene glycol and caution must be exercised in prescribing large scheduled doses of lorazepam for sedation of sick neonates.
Dermal exposure to a variety of chemical agents, including isopropyl alcohol, may be hazardous to the newborn (87,88). Toxic epidermal necrolysis was reported in a neonate after prolonged exposure to a commonly used distillate-containing adhesive remover (89). Unintended percutaneous absorption of toxic substances through the permeable skin of neonates has recurred with several substances. This has led to toxicity from methanol (90), isopropanol (91,92), hexachlorophene (93), iodine-containing topical disinfectants (94,95), aniline dye in diapers (96), and topical antibiotics such as neomycin (97). Advantageous and disadvantageous transcutaneous drug absorption by newborns has been reviewed (98). Chemical intoxication of the fetus and newborn has been reviewed in detail (70).
Inadvertent exposure of the neonate to a variety of chemicals may occur with little notice. Phthalate plasticizers accumulate in the myocardial and gastrointestinal tissue of neonates with umbilical catheters and those who receive blood products (99). Although phthalates probably produce minimal acute toxicity, they accumulate in tissues and may exert effects that are not recognized. Since the report by Hillman and colleagues (99), the extent of exposure of NICU patients to plastic products appears to have increased, causing greater exposure to soluble chemicals in the plastic, such as phthalates.
SPECIFIC CLASSES OF DRUGS
Antiarrhythmics
Treatment of cardiac arrhythmias has advanced with increased understanding of the transmembrane ion currents that control both normal and abnormal myocardial depolarization (100,101,102). Developmental changes in these channels are described using both electrophysiological techniques and molecular biological means of investigation (103). Careful interpretation of various studies is needed, however, as a result of species differences in the timing of innervation and in the development of various channels (103,104).
Antiarrhythmic drugs have been classified and reclassified according to the mechanisms responsible for arrhythmias, including effects on ion channels, duration of repolarization, and receptor interaction (Table 56-4) (100,105,106,107). A discussion of the various subtypes
of ion channels is beyond the scope of this chapter (100,101,102,108,109). Such classification systems help with analysis of the effects of individual antiarrhythmic drugs and selection of treatment for specific arrhythmias, but drugs and their metabolites may interact with multiple ion channels and more than one antiarrhythmic mechanism (106,110). Drugs within a given class, however, may differ in their effectiveness for an individual patient’s arrhythmia. Dosages for drugs used frequently for resuscitation of newborns are listed (see Table 56-5) (111).
of ion channels is beyond the scope of this chapter (100,101,102,108,109). Such classification systems help with analysis of the effects of individual antiarrhythmic drugs and selection of treatment for specific arrhythmias, but drugs and their metabolites may interact with multiple ion channels and more than one antiarrhythmic mechanism (106,110). Drugs within a given class, however, may differ in their effectiveness for an individual patient’s arrhythmia. Dosages for drugs used frequently for resuscitation of newborns are listed (see Table 56-5) (111).
TABLE 56-4 ANTIARRHYTHMIC DRUGS BY MECHANISM OF ACTION | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Drug selection for treatment of arrhythmias should be guided, whenever possible, by more precise identification of the type of arrhythmia. The most frequent arrhythmia requiring management in neonates is supraventricular tachycardia (SVT) (112). In general, SVT arises from either an aberrant conduction pathway or abnormal automaticity in a discrete focus that captures the ventricle (112). Reentrant mechanisms are the most common cause of SVT in neonates and usually involve retrograde conduction from the ventricle via the accessory conduction pathway to the atrium which then depolarizes prematurely. This has been designated orthodromic reciprocating tachycardia (ORT). A variant of ORT arises from a posterior accessory conducting pathway that causes a slower, but incessant form of SVT, permanent junctional reciprocating tachycardia (PJRT). PJRT, left untreated, may cause a cardiomyopathy and CHF. In the Wolff-Parkinson-White syndrome (WPW), the accessory conduction pathway can be identified during sinus rhythm from early depolarization that creates a beginning shoulder (delta wave) on the QRS complex with a shortened PR interval (112). Unfortunately, almost 50% of reentrant SVT in infants does not show preexcitation overtly during sinus rhythm. When the depolarization returns from the ventricle through the atrioventricular node to cause SVT, it is termed antidromic reentrant tachycardia, and causes a wide complex tachycardia. Reentrant tachycardias, either overt or concealed, can be identified during electrophysiologic studies by being able to be initiated and terminated by atrial or ventricular extrastimulation (110,112).
Nonreentrant SVT is rare in infancy and difficult to treat pharmacologically. SVT caused by a nonsinus node focus of abnormal automaticity is designated Automatic Atrial Tachycardia (AAT) and is difficult to treat medically (110). AAT must be distinguished from sinus tachycardia because the heart rate may gradually speed up and slow down. The p-wave morphology during AAT differs from that during sinus rhythm and the PR interval of conducted beats may lengthen, rather than shorten as the rate increases (110). SVT may also arise from a site of abnormal automaticity within or near the AV node, designated junctional ectopic tachycardia (JET) (110). JET is also quite difficult to treat with the usual antiarrhythmics (113,114). Treatment of SVT may be required in two situations, acute termination
of a symptomatic tachycardia and chronic suppression of SVT that has recurred or is likely to recur. Synchronized direct current electrocardioversion terminated SVT in about 50% of cases, but was more successful if P waves were not present (115). Vagal maneuvers, such as stimulation of the diving reflex through ice applied to the face, are still frequently used for the acute termination of SVT, although bilateral carotid massage or pressure on the eyes should be avoided.
of a symptomatic tachycardia and chronic suppression of SVT that has recurred or is likely to recur. Synchronized direct current electrocardioversion terminated SVT in about 50% of cases, but was more successful if P waves were not present (115). Vagal maneuvers, such as stimulation of the diving reflex through ice applied to the face, are still frequently used for the acute termination of SVT, although bilateral carotid massage or pressure on the eyes should be avoided.
TABLE 56-5 DRUGS FOR NEWBORN RESUSCITATION AND ACUTE TREATMENT OF ARRHYTHMIAS | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
For the acute treatment of SVT, adenosine has been used with moderate success. Adenosine slows spontaneous heart rate, prolongs the PR interval, and decreases the slope of phase 4 repolarization through activation of A1-purinoceptors coupled to sarcolemmal potassium channels. In one report, electroconversion was not required for conversion of infant onset SVT during the 6 years after the introduction of adenosine (116). The ventricle is minimally affected by adenosine whose action begins within seconds of administration. With a 9-second half-life, it must be administered as a rapid i.v. infusion over seconds, but it has been administered successfully by intraosseus infusion (117). Its short half-life limits its usefulness to the acute treatment of SVT. Adenosine treatment may be started with 50 to 150 μg/kg doses, with increases in the dose by 50 μg/kg every minute to a maximum of 300 to 350 μg/kg (106,118,119). Because theophylline is a competitive antagonist of adenosine (120), higher adenosine dosages may be required in infants treated with theophylline (121). Adenosine may precipitate bronchoconstriction and wheezing and hypotension as a result of vasodilatation (122).
SVT that has not responded to adenosine may be managed with an infusion of procainamide, a class 1A drug
that blocks both sodium and potassium channels (106). Rapid infusion may precipitate hypotension, whereas chronic treatment produces antinuclear antibodies in 50% to 90% of patients associated with a systemic lupus erythematosus-type syndrome (106,122). High concentrations of procainamide may depress myocardial contractility and predispose to congestive heart failure (106). Although more than half of a procainamide dose is excreted unchanged, it is metabolized in the liver to N-acetylprocainamide (NAPA), an active metabolite which acts through a different (class III) antiarrhythmic mechanism (106). Acetylation to NAPA involves the N-acetyltransferase enzyme system, which is involved in the metabolism of isoniazid. Adults may be phenotyped as fast or slow acetylators (123), however infants between birth and 2 months of age are uniformly slow acetylators as a result of the immaturity of the enzyme system (16). Both NAPA and procainamide accumulate with renal insufficiency. Procainamide levels of 4 to 8 mcg/ml are generally effective for control of arrhythmias.
that blocks both sodium and potassium channels (106). Rapid infusion may precipitate hypotension, whereas chronic treatment produces antinuclear antibodies in 50% to 90% of patients associated with a systemic lupus erythematosus-type syndrome (106,122). High concentrations of procainamide may depress myocardial contractility and predispose to congestive heart failure (106). Although more than half of a procainamide dose is excreted unchanged, it is metabolized in the liver to N-acetylprocainamide (NAPA), an active metabolite which acts through a different (class III) antiarrhythmic mechanism (106). Acetylation to NAPA involves the N-acetyltransferase enzyme system, which is involved in the metabolism of isoniazid. Adults may be phenotyped as fast or slow acetylators (123), however infants between birth and 2 months of age are uniformly slow acetylators as a result of the immaturity of the enzyme system (16). Both NAPA and procainamide accumulate with renal insufficiency. Procainamide levels of 4 to 8 mcg/ml are generally effective for control of arrhythmias.
Lidocaine, a class 1B antiarrhythmic, primarily blocks the fast inward sodium channel, like other local anesthetic antiarrhythmic drugs (106,122). Its effects are much greater on the conducting system and the ventricular muscle than on the atrium. It has active metabolites that also block the sodium channel. Plasma concentrations should be maintained between 2 and 5 μg/mL, because concentrations exceeding 6 μg/mL may produce seizures or respiratory arrest (106,122). Drugs known to decrease hepatic blood flow, such as cimetidine, will decrease hepatic clearance of lidocaine and increase concentrations unless the dose is reduced.
Chronic treatment of SVT is not needed in every patient (110). If the frequency of SVT is rare, not associated with cardiovascular compromise, and is easily terminated with vagal maneuvers, chronic treatment may not be required provided that the child can be monitored adequately at home (110). The presence of WPW, however, is a significant predictor of recurrence of SVT after stopping medications (124). Digoxin treatment of WPW is controversial, because it has been associated with sudden death in 1% to 5% of patients (110,125). In another series of patients with SVT, digoxin treatment was successful in 65% regardless of whether or not they showed preexcitation (116). A review of patients treated for SVT at Texas Children’s Hospital revealed no difference in the success rate for treatment of SVT with digitalis among patients with and without WPW (115).
Propranolol is frequently used to treat neonates with SVT, if digoxin is contraindicated because of the presence of WPW (106). It has special pharmacokinetic features that must be considered in its administration. When administered enterally, propranolol is cleared largely through first-pass extraction by the liver. It is metabolized extensively to an active metabolite, 4-hydroxypropranolol (126). During repetitive doses, hepatic extraction decreases, and plasma concentrations may vary widely at steady-state. If administered i.v., the propranolol dose must be reduced at least tenfold because it bypasses hepatic extraction and produces a dose- and concentration-related decrease in heart rate and cardiac contractility. Propranolol has been used most frequently to treat supraventricular and ventricular arrhythmias. It also has a role in treatment of sinus tachycardia related to hypermetabolic states such as thyrotoxicosis (122). Data on dosing, kinetics, and efficacy in neonates for other beta-adrenergic blocking drugs such as atenolol, nadolol, sotalol, and esmolol are limited.
Verapamil has been used for treatment of SVT with moderate success, but its use in infants younger than 12 months of age has been discouraged (106,127). As a result of its negative inotropic activity and ability to decrease sinus function, verapamil should not be used with beta-adrenergic blocking drugs in infants and children (122). They have been used together successfully in adults.
Amiodarone is a benzofuran compound with structural similarity to thyroxine that was originally synthesized in 1962 and released in the United States in 1985. Because of the associated toxicities, it was approved as a “last resort” drug that should be reserved for refractory, life-threatening arrhythmias (128). During the last few years, much more information has accumulated about both its efficacy and its toxicity. Amiodarone illustrates the limitations of a classification system for antiarrhythmics. It inhibits both the fast sodium channel and the slow calcium channel, has noncompetitive antisympathetic effects, and modulates thyroid function (129). Amiodarone has a relatively low negative inotropic effect and therefore may be the preferred antiarrhythmic treatment for infants and children with depressed myocardial function as a result of incessant tachycardias or perioperative tachycardias following surgery for congenital heart disease (130). Acutely, amiodarone slows AV node conduction with little effect on the QTc, although during chronic therapy, it lengthens QTc and prolongs the refractory period (129). Although reports are limited in neonates, the following adverse effects have been observed after amiodarone treatment of infants and children: photosensitivity (131,132), corneal deposits (132), gray skin color (132), abnormal liver function tests (132), hypothyroidism (131,132), hyperthyroidism (131,132), accelerated bone maturation (131), delayed longitudinal growth (131), excess weight gain (131), headaches (131), pulmonary infiltrates (133), and sleep disturbances (132). Amiodarone increases the concentration of several drugs, usually by decreasing clearance and volume of distribution, including digoxin, quinidine, procainamide, phenytoin, and flecainide. Decreased clearance of warfarin by amiodarone may precipitate hemorrhage. Overall, the frequency of adverse effects is greater in adults than in children and greater in older children than in infants.
Amiodarone treatment of refractory and serious arrhythmias in infants and children has been successful with limited acute toxicity. In a multi-center trial, Perry reported a 93% rate of improvement with the compassionate use of amiodarone for life-threatening arrhythmias in 40 patients from 8 centers (134). Several serious and refractory arrhythmias have been successfully treated with amiodarone, including: multifocal atrial tachycardia
(chaotic atrial tachycardia) (135,136), ventricular tachycardia as a result of intracardiac tumor (137), and refractory SVT (138,139). In a study by Burri and associates, i.v. amiodarone was reported to be safe and effective therapy for life-threatening incessant tachycardias in infants (130). Twenty-three hemodynamically unstable infants (median age 8 days) with life-threatening tachycardias (17 supraventricular, 6 ventricular) were treated with i.v. amiodarone 5 mg/kg administered over 1 hour followed by 5 μmg/kg/min with stepwise increase to 25 μg/kg/min (130). Amiodarone was effective in 83% of infants. Med ian time to arrhythmia control was 24 h and the median maintenance dose was 15 μg/kg/min (range 5-26 μg/kg/min). Adverse effects were not significant: sinus bradycardia in 2 patients and hypotension in one infant, which resolved after dose reduction. Choreoathetotic movements and elevated liver enzymes occurred in two patients.
(chaotic atrial tachycardia) (135,136), ventricular tachycardia as a result of intracardiac tumor (137), and refractory SVT (138,139). In a study by Burri and associates, i.v. amiodarone was reported to be safe and effective therapy for life-threatening incessant tachycardias in infants (130). Twenty-three hemodynamically unstable infants (median age 8 days) with life-threatening tachycardias (17 supraventricular, 6 ventricular) were treated with i.v. amiodarone 5 mg/kg administered over 1 hour followed by 5 μmg/kg/min with stepwise increase to 25 μg/kg/min (130). Amiodarone was effective in 83% of infants. Med ian time to arrhythmia control was 24 h and the median maintenance dose was 15 μg/kg/min (range 5-26 μg/kg/min). Adverse effects were not significant: sinus bradycardia in 2 patients and hypotension in one infant, which resolved after dose reduction. Choreoathetotic movements and elevated liver enzymes occurred in two patients.
Dosages of amiodarone have varied widely from study to study. Initial treatment with amiodarone usually begins with a loading dose (5 mg/kg) followed by an infusion of 10 to 15 mg/kg/day (134) to 7.5 to 13.5 mg/kg/day for 9 to 10 days (140). Larger dosages were required for infants less than 1 year of age in one study (132). Oral treatment alone has been successful in some studies (132,141). In a study by Etheridge and associates, 44 neonates and infants (1.0 =6μ 1.5 months) with difficult to control tachycardias were treated with oral amiodarone using a 7- to 10-day load of amiodarone at either 10 or 20 mg/kg/day in two divided doses (141). The higher loading dose was used for more difficult to control tachycardias. Oral propranolol (2 mg/kg/d) was added for sustained or symptomatic episodes of tachycardia (n =5μ 25). Rhythm control was achieved in all patients. Infants were discharged on maintenance amiodarone dose of 5 to 10 mg/kg/day (mean dose 7 =6μ 2 mg/kg/day) and drugs were discontinued as tolerated. Sixty-eight percent of patients remained free of arrhythmias at one year, despite discontinuation of propranolol and amiodarone. There were no side effects necessitating drug withdrawal.
The prolonged loading dose likely relates to amiodarone’s extremely long half-life that has been estimated in adults to exceed 50 days (128). Levels of amiodarone are kept between 0.8 and 2.0 mg/L in adults (132), but there is no clear relationship between levels of amiodarone or its metabolite desethylamiodarone and toxicity or efficacy (142,143).
Newborns with fetal arrhythmias may be treated with amiodarone and require evaluation of potential adverse effects in the nursery. A healthy fetus whose mother was treated with amiodarone for sick sinus syndrome delivered and breast fed with no changes in thyroid function (144). Both neonatal hyperthyroidism and hypothyroidism with or without a goiter have been observed after fetal exposure to amiodarone (145,146,147,148).
Anticonvulsants
Seizures remain a frequent therapeutic problem for neonates admitted to an intensive care unit. They may be caused by a variety of disorders, such as meningitis, inadequate pyridoxal-5-phosphate binding, i.e., pyridoxine dependency, hypoglycemia, hypocalcemia, inborn errors of metabolism, neonatal abstinence from narcotics, or intracranial hemorrhage (149). Because treatment differs according to etiology, causes for seizures outside the brain should always be considered and evaluated. Some noncerebral causes of seizures may produce recurrent seizures that require prolonged anticonvulsant drug treatment, in addition to treatment of the primary metabolic or infectious disorder. Because prolonged seizures per se may harm the brain, treatment should not be delayed (149).
Phenobarbital remains the mainstay of treatment and is effective in approximately one-third of cases; babies who respond tend to have a smaller seizure burden and a rela tively normal background electroencephalogram (150,151). Phenobarbital is often ineffective as a first line anticonvulsant in neonates with seizures in whom the background electroencephalogram (EEG) is significantly abnormal (151,152). Although seizures are controlled in some patients with phenobarbital levels of 15 μg/mL, the minimal effective therapeutic concentration should be regarded as 20 μg/mL and many patients may require concentrations as high as 40 μg/mL to achieve seizure control (153). As a result of a volume of distribution of approximately 0.9 L/kg in neonates, a loading dose of 15 to 20 mg/kg is required to reach a minimum concentration of 20 μg/mL (154). A total loading dose of up to 40 mg/kg rapidly achieves therapeutic levels (150). Caution is advised in nonventilated infants because a single loading dose of 40 mg/kg can result in apnea (150). Accordingly, splitting the loading dose into two 20 mg/kg doses is advised.
The half-life of phenobarbital is prolonged at birth, ranging from 43 to 217 hours (155). Consequently, lower doses of phenobarbital (2-3 mg/kg/day) should be used during the first week of life. Phenobarbital clearance increases significantly during the neonatal period and the half-life decreases to approximately 45 hours at 28 days of life (154). Thus, maintenance doses will often need to be increased within the first several weeks after birth (3-5 mg/kg/day). A good correlation has been demonstrated between plasma and brain concentrations of both phenobarbital and phenytoin, so that increasing plasma concentrations should increase the anticonvulsant concentration at the site of action (12).
Concerns have increased about the effects of long-term phenobarbital treatment on cognitive development. When phenobarbital was compared to placebo in two randomized groups of children treated for 2 years for febrile seizures, the phenobarbital-treated group had significantly lower intelligent quotients (IQs) (156). In a randomized, crossover trial of three anticonvulsants, phenobarbital produced more impairment of neuropsychological performance than did carbamazepine or phenytoin (157). The American Academy of Pediatrics (AAP) Committee on Drugs summarized concerns about the cognitive effects of phenobarbital (158).
Phenytoin has served as the second line anticonvulsant for neonates. Phenytoin and phenobarbital have been
shown to be equally but incompletely effective as anticonvulsants in neonates.(151) The severity of the seizure is a stronger predictor of successful treatment than the agent. Painter and associates evaluated phenytoin (15 mg/kg) as an alternative to phenobarbital. Of the 29 babies administered phenytoin as first-line treatment, 45% responded (151). Phenytoin and lidocaine are probably more effective than any of the benzodiazepines as second line, but very few evaluation studies have been reported (150). Disadvantages of phenytoin are its highly variable and nonlinear pharmacokinetics, i.e., exponential elimination decreases with increasing serum concentrations. The latter phenomenon requires considerable caution because small increases in maintenance dose may result in disproportionately large increases in plasma concentrations. The initially prolonged half-life of phenytoin decreases rapidly during the neonatal period from 57.3 =6μ 48.2 hours during the first days of life to 19.7 =6μ 1.3 hours during the fourth week (33). In term infants between 2 and 21 weeks of age, the half-life ranged from 6.6 hours to 15.1 hours (20). However, preterm infants often exhibit prolonged half-lives (15.6 to 160 hours) up to 18 days after birth (20).
shown to be equally but incompletely effective as anticonvulsants in neonates.(151) The severity of the seizure is a stronger predictor of successful treatment than the agent. Painter and associates evaluated phenytoin (15 mg/kg) as an alternative to phenobarbital. Of the 29 babies administered phenytoin as first-line treatment, 45% responded (151). Phenytoin and lidocaine are probably more effective than any of the benzodiazepines as second line, but very few evaluation studies have been reported (150). Disadvantages of phenytoin are its highly variable and nonlinear pharmacokinetics, i.e., exponential elimination decreases with increasing serum concentrations. The latter phenomenon requires considerable caution because small increases in maintenance dose may result in disproportionately large increases in plasma concentrations. The initially prolonged half-life of phenytoin decreases rapidly during the neonatal period from 57.3 =6μ 48.2 hours during the first days of life to 19.7 =6μ 1.3 hours during the fourth week (33). In term infants between 2 and 21 weeks of age, the half-life ranged from 6.6 hours to 15.1 hours (20). However, preterm infants often exhibit prolonged half-lives (15.6 to 160 hours) up to 18 days after birth (20).
Phenytoin is a water-insoluble base and the commercially available injection formulation is prepared using 40% propylene glycol at a final pH of 12. Side effects associated with parenteral phenytoin, caused primarily by its high pH and the propylene glycol content, include burning at the infusion site and severe local cutaneous reactions following infiltration into surrounding tissue (159). Many of these side effects can be avoided in children with the use of fosphenytoin, a phosphorylated prodrug of phenytoin which has few cardiac and local irritation adverse effects (160). The accepted therapeutic serum concentration range for phenytoin (or fosphenytoin converted to phenytoin) is 10 to 20 μg/mL and a loading dose of 15 to 20 mg/kg will achieve a therapeutic concentration (161). To avoid hypotension, arrhythmias, and precipitation during administration, phenytoin should be administered slowly (maximum 0.5-1 mg/kg/min) into an i.v. line containing only saline. Because the drug is caustic to tissues, intramuscular administration should be avoided and intravenous doses should be infused through as large a vein as is available taking care to avoid extravasation.
Interpretation of phenytoin plasma concentrations requires consideration of protein binding. In adults, the unbound concentration of phenytoin is approximately 10% of the total concentration (162). Thus, the therapeutic range for unbound (free) phenytoin is 1 to 2 μg/mL. Because plasma protein concentrations and binding affinity of albumin are low in newborns, plasma protein binding of phenytoin is reduced in infants. In normobilirubinemic newborns, the free (unbound) fraction of phenytoin is 15% to 20% and in hyperbilirubinemic infants, the free fraction may reach 30% (20,163,164). Thus, despite apparent subtherapeutic total concentrations (=,μ10 μg/mL) free concentrations may be within the therapeutic range of 1 to 2 μg/mL. Monitoring free phenytoin concentrations may be useful in this circumstance to determine if concentrations are within the therapeutic range. If free concentrations of phenytoin are not available, targeting total concentrations of 6 to 14 μg/mL are appropriate in the immediate neonatal period (20). However, as protein binding normalizes in the weeks to months after birth, the therapeutic range will require readjustment to 10 to 20 μg/mL.
As a result of the significant variability in pharmacokinetics during the first weeks of life, a standard dose of phenytoin cannot be recommended and plasma concentrations must be monitored. During the first week of life, doses of 4-8 mg/kg/day may be required (20). However, chronic oral phenytoin therapy in infants often requires much higher doses (161). By the second week after birth, most infants will require at least 8 mg/kg/day divided every 6 to 8 hours (20), however oral doses as high as 17.7 =6μ 4.3 mg/kg/day and intravenous doses of 25 mg/kg/day have been required to maintain therapeutic concentrations (161,165). Although some have attributed these dose requirements to poor absorption of the oral formulation (161), high doses are also required after intravenous administration (165). Other investigators have demonstrated complete absorption in infants receiving oral doses (166). High oral dosage requirements in infants likely reflects several pharmacokinetic events, including poor and erratic absorption, increased phenytoin metabolism during the first year of life, and hepatic enzyme induction by concomitant treatment with phenobarbital (161,165,166).
Recently, the introduction of fosphenytoin has significantly reduced the potential for the adverse effects usually associated with phenytoin, including pain on administration, extravasation injury, propylene glycol-associated hyperosmolality (83), CNS changes (85,86), acidosis (167), and cardiovascular compromise (168) following rapid intravenous injection (159). Fosphenytoin is a water-soluble prodrug that is rapidly hydrolyzed to phenytoin via blood and tissue phosphatases (169). Because it is an aqueous solution and contains no propylene glycol, it has few pharmaceutical compatibility problems and may be administered by intravenous injection or by intramuscular injection (169). Because fosphenytoin is converted to phenytoin, issues regarding protein binding, therapeutic concentrations, and drug interactions associated with phenytoin must still be considered. There is limited published experience of its use in newborn infants (160).
Benzodiazepines can be used for additional drug therapy beyond phenobarbital and phenytoin to control seizures in the newborn. Diazepam has been used to treat refractory seizures in neonates, but its efficacy is limited by its short duration of action, respiratory depression, prolonged half-life, and metabolism to active metabolites (170). Lorazepam, in a dose of 0.05 to 0.1 mg/kg has been effective for controlling refractory seizures in neonates and has a duration of action up to 24 hours (84,170). Total doses up to 0.15 mg/kg may be necessary to control neonatal seizures that are refractory to phenobarbital (171). Because metabolism of lorazepam requires glucuronidation, which is poorly developed in newborns, the half-life
of lorazepam in newborns is prolonged (mean 40 hours) and correlates with gestational age (172). The dose of benzyl alcohol associated with lorazepam treatment is low enough not to represent a risk for toxicity (173). Some have reported “seizure-like” stereotypic movements after lorazepam administration (174,175). The cause of this movement disorder is not known.
of lorazepam in newborns is prolonged (mean 40 hours) and correlates with gestational age (172). The dose of benzyl alcohol associated with lorazepam treatment is low enough not to represent a risk for toxicity (173). Some have reported “seizure-like” stereotypic movements after lorazepam administration (174,175). The cause of this movement disorder is not known.
Recently midazolam has been used in adults and children for treatment of epileptic episodes of various types, including status epilepticus (176,177). A study was conducted in six neonates (aged 1-9 days; gestation, 30-41 weeks) with persistent seizures despite a mean phenobarbital concentration of 50.5 mg/L (178). Seizures were controlled in all patients within one hour of administering continuous intravenous infusions of midazolam (0.1-0.4 mg/kg/hour). Because neonatal seizures are sometimes refractory to high-dose phenobarbital, midazolam may be a valuable adjunctive therapy (178). Care must be taken to avoid rapid bolus doses of midazolam over 1 to 2 minutes that may cause hypotension in infants receiving fentanyl infusions (15) or tonic-clonic movements similar to those reported in older children following lorazepam (179).
Neonatal seizures refractory to phenobarbital have recently been shown to respond to oral administration of carbamazepine (180). Carbamazepine is a first-line anticonvulsant in the treatment of partial seizures with or without secondary generalization in children and adults. Ten preterm infants (gestational age 23 to 34) with neonatal seizures refractory to phenobarbital (plasma concentration ranging from 50-64 mg/L) received 7-15 mg/kg/day in 2 to 3 divided doses to achieve a plasma concentration of 3-12 mg/L. Nine out of the ten patients had excellent results without adverse effects. In another report, 10 full-term neonates with two or more seizures as a result of hypoxic-ischemic encephalopathy were administered 10 mg/kg of carbamazepine as a loading dose (via nasogastric tube) followed by a maintenance regimen of 15 to 21 mg/kg/day (181). Absorption of carbamazepine was excellent and therapeutic levels were reached in 2 to 4 hours with peak levels achieved in 9.2 ± 4.2 hours. The elimination half-life was 24.5 hours. Levels dropped precipitously around 8 to 15 days and declined slowly over the next 3 months. Seizure control was excellent and there were no adverse effects (181).
Of the newer anticonvulsants, lamotrigine is effective in adults and children with generalized and partial seizures. Lamotrigine is a phenyltriazine structurally unrelated to conventional antiepileptic drugs (AEDs). Recently, lamotrigine has been shown to be a useful agent for treatment of refractory seizures, partial seizures, and infantile spasms (182,183). Thirteen infants (=,μ1 year) with a mean age of 3.7 months (five were 2-4 weeks) had either partial seizures or infantile spasms or both (182). Both were significantly reduced (p < 0.05) and lamotrigine was well tolerated. The mean apparent clearance was 82% lower in the infants less than 2 months of age compared with those that were 2 to 12 months of age. Lamotrigine is mainly eliminated by hepatic metabolism to an inactive metabolite through glucuronidation (184). Co-medication with enzyme inducers increases metabolism (184), and although not studied in neonates, one would expect a drug interaction with phenobarbital. The adverse effect which often leads to drug discontinuation is skin reactions. Overall, rash develops in 12% of patients and the risk of rash is approximately 3 times greater in children as compared to adults (184). Other reported adverse effects include leucopenia, agranulocytosis, and hepatic failure.
Topiramate, another one of the newer antiepileptic drugs, is currently approved for adjunctive therapy for the treatment of generalized tonic-clonic seizures and partial seizures, and Lennox-Gastaut syndrome. It differs both structurally and pharmacologically from other classes of AEDs. Topiramate is a sulfamate-substituted monosaccharide D-fructose derivative with limited protein binding (9%-17%) (185). It is primarily eliminated by renal excretion with a small portion metabolized by hydrolysis, hydroxylation, and glucuronidation (185). No studies have been conducted in neonates, although a report of 5 infants (4 term and one born at 36 weeks) exposed to topiramate in utero indicated extensive transplacental transfer and a half-life of approximately 24 hours, which is close to the 20 to 30 hours reported in adults (185).
The decision to continue or to stop treatment with anticonvulsant medications before discharge from the NICU remains controversial. Scher and Painter suggest that anticonvulsants may be discontinued for infants who show no abnormalities of the brain on imaging studies, have an age-appropriate neurologic examination, and have a normal interictal electroencephalogram (186). Up to 30% of neonates with seizures later have epilepsy, but frequently the later seizure pattern takes the form of infantile spasms or minor motor seizures that are not very responsive to phenobarbital and phenytoin (186).
Antihypertensives
The relationship between blood pressure, gestational age, birth weight and postconceptional age was defined by Zubrow and associates in 695 neonates studied over a period of 3 months (187). Based on these data, hypertension in term and preterm infants may be defined as blood pressure above the upper limit of the 95% confidence interval for infants of similar gestational or postconceptual age and size (188). Drug treatment for hypertension should start with a single drug from one of the following classes, angiotensin-converting enzyme (ACE) inhibitors, vasodilators, beta-receptor antagonists, calcium channel blockers, or diuretics (189).
For severe hypertension in the neonate, a continuous intravenous infusion is advantageous because blood pressure fluctuations observed with intermittent dosing are eliminated and the dose may be titrated to keep blood pressure within narrow parameters. Antihypertensive agents that have been used in neonates or infants by continuous infusion include esmolol (190), nitroprusside, and nicardipine (191,192). Small studies have found that
intravenous nicardipine may be effective in treating hypertensive neonates (191,192). Milou and associates studied 20 neonates (15 preterm) who were administered intravenous nicardipine 0.5 μg/kg/min (maximum 0.74 ± 0.41 μg/kg/min) for 14.6 ± 11.6 days (192). Blood pressure significantly decreased (approximately 20%) after 3, 6, 12, 24, and 48 hours of nicardipine treatment. No hypotension was observed. Gouyon et al. studied 8 preterm infants with systemic hypertension and treated them with intra venous nicardipine 0.5 μg/kg/min (n =2) or 1.7 μg/kg/min (n =6) (191). The maximum infusion rate was 1.56 ± 0.45 μg/kg/min and the treatment duration was 15.9 ± 10.3 days. Systolic blood pressure significantly decreased after 12 and 24 hours of nicardipine treatment (-17 ± 17% and -21 ± 10%, respectively) and diastolic blood pressure decreased after 24 hours of treatment (-22 ± 16%) No hypotension or other side effects were noted. Hypertensive emergencies are infrequent in neonates, but can be treated with esmolol, hyralazine, labetalol, sodium nitroprusside if renal function is normal, fenoldopam, isradipine, and minoxidil (189). As observed in older patients, thiocyanate and cyanide may accumulate during sodium nitroprusside treatment. Severe prolonged hypotension has been reported in severely hypertensive neonates treated with enalaprilat (189).
intravenous nicardipine may be effective in treating hypertensive neonates (191,192). Milou and associates studied 20 neonates (15 preterm) who were administered intravenous nicardipine 0.5 μg/kg/min (maximum 0.74 ± 0.41 μg/kg/min) for 14.6 ± 11.6 days (192). Blood pressure significantly decreased (approximately 20%) after 3, 6, 12, 24, and 48 hours of nicardipine treatment. No hypotension was observed. Gouyon et al. studied 8 preterm infants with systemic hypertension and treated them with intra venous nicardipine 0.5 μg/kg/min (n =2) or 1.7 μg/kg/min (n =6) (191). The maximum infusion rate was 1.56 ± 0.45 μg/kg/min and the treatment duration was 15.9 ± 10.3 days. Systolic blood pressure significantly decreased after 12 and 24 hours of nicardipine treatment (-17 ± 17% and -21 ± 10%, respectively) and diastolic blood pressure decreased after 24 hours of treatment (-22 ± 16%) No hypotension or other side effects were noted. Hypertensive emergencies are infrequent in neonates, but can be treated with esmolol, hyralazine, labetalol, sodium nitroprusside if renal function is normal, fenoldopam, isradipine, and minoxidil (189). As observed in older patients, thiocyanate and cyanide may accumulate during sodium nitroprusside treatment. Severe prolonged hypotension has been reported in severely hypertensive neonates treated with enalaprilat (189).
Intermittently administered intravenous agents may be useful for patients with mild-to-moderate hypertension. These agents include hydralazine, labetalol, and enalaprilat, (193) although enalaprilat should be used with great caution as a result of the potential for significant and prolonged hypotension (188).
Oral antihypertensives may be used in infants with mild-to-moderate hypertension or in patients being transitioned from intravenous therapy. Angiotensin-converting enzyme inhibitors (e.g., captopril) have a special role in the treatment of neonatal renovascular hypertension that is caused by markedly elevated renin and angiotensin (194). Aortic catheters may distribute microemboli to the kidneys, which increase secretion of renin and angiotensin in response to focal underperfusion. Although captopril may be relatively specific treatment for many cases of renovascular hypertension in neonates, dosage adjustments are difficult because a liquid dosage form is not commercially available. An extemporaneously compounded dosage form containing ascorbate has been shown to be stable for 14 days at room temperature (195). Captopril treatment of neonates should start with doses of 0.01 mg/kg, rather than the earlier recommendations of 0.1 to 0.3 mg/kg/dose, to avoid hypotension and acute renal insufficiency (196). Doses should be increased daily if hypertension persists. Initial captopril treatment may cause a triphasic reaction of hypotension with renal failure, managed by dose reduction, followed by a rise in blood pressure despite increasing doses above the original dose (197). At the start of captopril treatment, hypotension is exaggerated in salt and water-depleted patients (198). As in adults, captopril may be useful for afterload reduction in the treatment of chronic congestive heart failure in newborns (198). Other oral agents that have been used include the vasodilators hydralazine and minoxidil, and the calcium channel blocker isradipine (188). Isradipine can be compounded into a suspension and its successful and safe use has been reported in infants and children (199,200). The use of isradipine suspension allows infants and young children to be treated as readily as older children.
Antimicrobials
Beta-Lactams
Historically, the selection of antibiotics for the treatment of sepsis and infections in neonates has been empiric based on the most frequent pathogens observed in the clinical setting. Ampicillin and an aminoglycosides have been the mainstay for treatment of presumed sepsis and meningitis in the newborn (201). The advantages of this combination include low cost, relatively few problems with resistance (202,203), and a combined spectrum of activity that covers the majority of neonatal pathogens, including Group B Streptococcus, E. coli, and Listeria monocytogenes (201). However, resistance to antimicrobial agents and changes in the pathogens causing neonatal infections must be the focus of ongoing surveillance to respond with the most appropriate antibiotic combination. As antepartum antibiotic use for the prevention of group B streptococcal disease increases, organisms recovered in early onset disease may shift toward Escherichia coli resistant to ampicillin (204,205). In a study of 5447 very-low-birth-weight (VLBW) infants born between 1998 and 2000, there was a marked reduction in group B streptococcal sepsis (from 5.9 to 1.7 per 1,000 live births, p < 0.001) and an increase in Escherichia coli sepsis (from 3.2 to 6.8 per 1,000 live births, p 0.004) as compared to VLBW infants born between 1991 and 1993 (205). Most E. coli isolates from the recent birth cohort (85%) were resistant to ampicillin. Ampicillin-resistant strains of Morganella morganii have also been reported to cause early onset sepsis in babies born to mothers who received intrapartum ampicillin (206).
The use of third-generation cephalosporins (e.g., cefotaxime, ceftriaxone, and ceftazidime) continues to proliferate, primarily as a result of their excellent penetration into cerebrospinal fluid, minimal nephrotoxicity, and broad gram-negative activity, including Klebsiella sp. and many gentamicin-resistant isolates (207). When L. monocytogenes or Enterococci are potential causes of infection, initial empiric therapy with third generation cephalosporins must be combined with ampicillin because the cephalosporins are not active against these pathogens. The choice of third generation cephalosporins over an aminoglycoside will depend on institutional susceptibility patterns. However, because excessive use of extended spectrum cephalo-sporins is associated with selection for highly resistant, extended spectrum β-lactamase (ESBL) producing organisms (208,209,210,211), many experts favor a regimen of a penicillin and aminoglycoside for primary therapy of suspected
sepsis (208). It has been proposed that the biliary excretion of broad spectrum antibiotics promotes overgrowth of resistant strains (especially Enterobacter and Enterococcus) which can translocate from the intestines into the bloodstream to causeinfection (212,213).
sepsis (208). It has been proposed that the biliary excretion of broad spectrum antibiotics promotes overgrowth of resistant strains (especially Enterobacter and Enterococcus) which can translocate from the intestines into the bloodstream to causeinfection (212,213).
Among the cephalosporins, there is considerable experience using cefotaxime in severe neonatal infections (214). Cefotaxime has often replaced gentamicin for coverage of gram-negative bacilli during outbreaks of sepsis caused by gentamicin-resistant gram-negative bacteria (209,215). Although this combination may increase the relative number of enterobacter isolates and their resistance (209,211, 216,217). The incidence of serious gram-negative infections decreased during a 5-year period when cefotaxime was substituted for gentamicin (215). Ceftriaxone has a very similar spectrum of activity as cefotaxime, with the primary difference being its pharmacokinetic profile. Ceftriaxone was well tolerated in 80 neonates (mean gestational age 34 weeks), although six patients had sonographic findings consistent with ‘biliary sludge’ which resolved spontaneously within 2 weeks (218). It is believed that ceftriaxone forms a complex with calcium that is poorly soluble in bile resulting in biliary sludge, which appears to be a transient phenomenon (219). Ceftriaxone may also displace bilirubin from albumin binding sites (220,221). An in vivo study demonstrated the potential significance of this interaction and the investigators recommended that ceftriaxone be withheld from jaundiced neonates or premature newborns with acidosis, hypoxia, or sepsis who are at increased risk for developing bilirubin encephalopathy (221).
Certain cephalosporins have unique properties that fit clinical problems encountered frequently in neonates, e.g., ceftazidime is considered the drug of choice to treat Pseudomonas infections. It should be combined with an aminoglycoside for synergy and to avoid emergence of resistant strains (222,223). A recent study of 1316 cases of suspected cases of sepsis in the newborn (median age ≤ 3 days) found that ceftazidime in combination with ampicillin had a significantly higher cure rate (97%) than ampicillin and an aminoglycoside (66%) (224). No problems with ceftazidime resistance were encountered. Pharmacokinetic data in preterm infants indicate that the ceftazidime dose during the first 2 weeks of life should be based on gestational age and postnatal age-related changes in glomerular filtration rate (225,226). Additionally, exposure of preterm infants to indomethacin or asphyxia in term infants may reduce ceftazidime’s clearance and dosage requirements (225).
The ureidopenicillins, ticarcillin and piperacillin, provide broader gram-negative coverage than ampicillin and are particularly suitable for the treatment of late-onset sepsis (227). For treatment of Pseudomonas sp. infections, piperacillin is more active in vitro than ticarcillin and may be combined with an aminoglycoside for synergy and to decrease the emergence of resistant organisms (228,229,230). When piperacillin is combined with an aminoglycoside such as gentamicin, the combination provides excellent activity against aerobic and anaerobic bacteria encountered after intestinal perforation. Piperacillin dosage requirements are based on both gestational and postnatal age (231).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


