Drug Action and Therapy in the Infant and Child
Ralph E. Kauffman
Infancy and childhood extend from 2 months of age to the onset of puberty, which typically occurs at approximately 10 to 12 years in girls and 12 to 14 years in boys. The first 2 to 3 years of life is a period of particularly rapid growth and development. Body weight doubles by 5 months and triples by the first birthday. Body length increases by 50% during the first year. Body surface area doubles by the first birthday. Caloric expenditure increases threefold to fourfold during the first year. The child becomes ambulatory, develops socialization, and learns verbal language.
Substantial changes in body proportions and composition accompany growth and development. Major organ systems differentiate, grow, and mature throughout infancy and childhood. Although growth and development are most rapid during the first several years of life, maturation continues at a slower pace throughout middle and later childhood. This dynamic process of growth, differentiation, and maturation is what sets the infant and child apart from adults, both physiologically and pharmacologically. It should be no surprise, then, that important changes in response to and biodisposition of drugs occur during infancy and childhood. These changes influence the response to, toxicity of, and dosing regimens for drugs.
This chapter focuses on the impact of growth and development on drug actions and biodisposition and the resultant practical implications for pharmacotherapy in children. Comparative pharmacologic data in children and adults are used to illustrate general principles.
Developmental Changes in Body Composition and Proportion
Developmental changes in body composition, body proportions, and relative mass of the liver and the kidneys affect pharmacokinetic characteristics of drugs at different ages. It is therefore important to review the relevant changes that take place between early infancy and pubescence.
The proportions of body weight contributed by fat, protein, and intracellular water, respectively, change significantly during infancy and childhood (Fig. 3.1). Total-body water constitutes approximately 75% to 80% of body weight in the full-term newborn. This decreases to approximately 60% by 5 months of age and remains relatively constant thereafter. Although the percentage of total-body weight constituted by total-body water does not change significantly after late infancy, there is a progressive decrease in extracellular water from infancy to young adulthood. In addition, the percentage of body weight contributed by fat doubles by 4 to 5 months of age, primarily at the expense of total-body water. During the second year of life, protein mass increases, with a compensatory reduction in fat. This corresponds to ambulation and loss of “baby fat” during the transition from infancy to childhood.
Liver and kidney size, relative to body weight, also changes during growth and development (1) (Fig. 3.2). These two organs reach maximum relative weight in the 1- to 2-year-old child, at the period of life when capacity for drug metabolism and elimination also tends to be greatest. Likewise, body surface area is greatest relative to body mass in the infant and the young child compared with the older child and the young adult (1,2) (Fig. 3.3).
Developmental Changes in Organ Function
Gastrointestinal Tract
The most common route of drug administration is the gastrointestinal (GI) tract. Therefore, physiologic and structural changes in the GI tract during development may alter both the rate and the extent to which a drug is absorbed. Clinically important developmental changes in the GI tract that may affect drug absorption occur predominantly during the newborn period, infancy, and early childhood. By school age, there is little discernible difference between children and adults.
A number of changes occur in the GI tract during early development that may affect drug absorption (3). Gastric acid production is decreased during infancy (4,5,6,7). Gastric
pH is neutral (6,7,8) at birth, drops during the first 24 hours, and increases toward neutral in the later newborn period. The capacity for gastric acid production is physiologically decreased during infancy and reaches adult levels sometime during the first 2 years of life. Gastric emptying is irregular and erratic during infancy and approaches adult patterns by 6 to 8 months of age (8). Gut motility in newborns and young infants is irregular with a pattern of peristaltic activity different from that in adults (9,10). This can lead to a longer transit time through the gut. Transit times in premature infants range from 8 to 96 hours, compared to 4 to 12 hours in adults. In the older infant and toddler, coordinated propulsive gut motility is more efficient, resulting in a transit time less than that in adults. Small gut surface area is greater in infants and young children relative to body mass than in adults (11). The gut in the infant is more permeable to large molecules than that of the older child (12). The infant gut has the capability of absorbing intact protein and high–molecular-weight drugs that are not absorbed by the gut of the older child and adult. Pancreatic lipase activity is decreased in newborns and during early infancy. First-pass uptake is presumed to be decreased in infancy, proportional to decreased transport and drug metabolism. Dietary habits of infants are quite different from those of older children and adults. They feed frequently, the diet consists of breast milk or formula for the first 6 months, and gastric contents are easily buffered. The combination of delayed gastric emptying and frequent feeds results in the presence of nutrients in the stomach for the majority of time between feedings.
pH is neutral (6,7,8) at birth, drops during the first 24 hours, and increases toward neutral in the later newborn period. The capacity for gastric acid production is physiologically decreased during infancy and reaches adult levels sometime during the first 2 years of life. Gastric emptying is irregular and erratic during infancy and approaches adult patterns by 6 to 8 months of age (8). Gut motility in newborns and young infants is irregular with a pattern of peristaltic activity different from that in adults (9,10). This can lead to a longer transit time through the gut. Transit times in premature infants range from 8 to 96 hours, compared to 4 to 12 hours in adults. In the older infant and toddler, coordinated propulsive gut motility is more efficient, resulting in a transit time less than that in adults. Small gut surface area is greater in infants and young children relative to body mass than in adults (11). The gut in the infant is more permeable to large molecules than that of the older child (12). The infant gut has the capability of absorbing intact protein and high–molecular-weight drugs that are not absorbed by the gut of the older child and adult. Pancreatic lipase activity is decreased in newborns and during early infancy. First-pass uptake is presumed to be decreased in infancy, proportional to decreased transport and drug metabolism. Dietary habits of infants are quite different from those of older children and adults. They feed frequently, the diet consists of breast milk or formula for the first 6 months, and gastric contents are easily buffered. The combination of delayed gastric emptying and frequent feeds results in the presence of nutrients in the stomach for the majority of time between feedings.
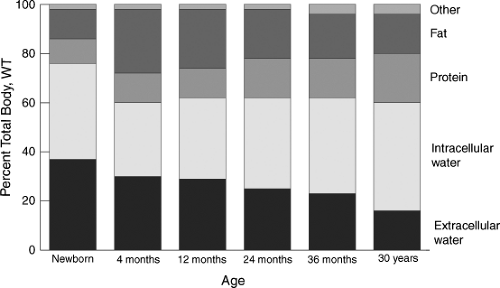 Figure 3.1. Change in proportional body composition during childhood. WT, Weight. (Adapted from Habersang RWO. Dosage. In: Shirkey HC, ed. Pediatric therapy, 6th ed. St. Louis, MO: Mosby, 1980:17–20, with permission.) |
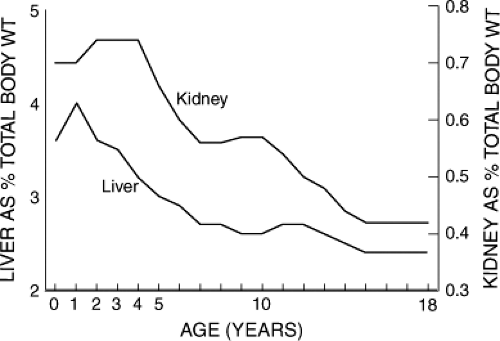 Figure 3.2. Change in relative liver and kidney mass expressed as percentage of body weight (WT) from infancy to young adulthood (1). |
Organs of Elimination
The liver and the kidney are the primary organs of drug metabolism and elimination. Although a great deal has been written about the immaturity of hepatic and renal function in the neonate, relatively less emphasis has been placed on maturation of the liver and the kidney during childhood and adolescence and the impact on drug disposition.
Hepatic function is complex, and metabolic pathways for various substrates develop at different rates. Bromsulphalein has been used as a substrate to evaluate maturation of hepatic clearance during infancy and childhood (13,14). Bromsulphalein clearance, normalized for body surface area, increases rapidly during the first 3 months of life, significantly exceeds adult clearance in the preschool child, and declines to adult levels during adolescence (Fig. 3.4). During the last decade a great deal has been learned about the development of drug-metabolizing enzymes, particularly the cytochromes P450, which are responsible for phase I metabolism of most drugs (15,16). The reader is referred to Chapter 4 for a detailed discussion of drug metabolism in the infant and child.
Glomerular filtration rate, as reflected by endogenous creatinine clearance, is physiologically decreased in the newborn but increases rapidly during the first year of life. Creatinine clearance, normalized for body surface area, equals adult clearance by 1 year of life, and there is some evidence that average clearance in prepubescent children exceeds clearance in adults (14,17,18) (Fig. 3.5). Tubular function matures later than glomerular function. However, tubular function is essentially mature by 1 year of age.
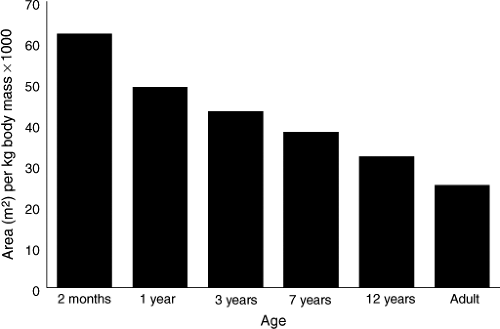 |
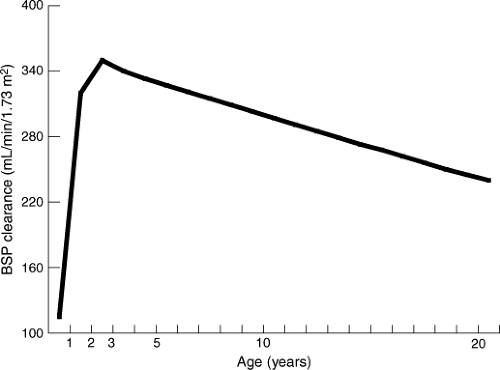 Figure 3.4. Change in hepatic clearance (expressed as mL/min/1.73 m2) of Bromsulphalein (BSP) during childhood. (Adapted from Habersang RWO. Dosage. In: Shirkey HC, ed. Pediatric therapy, 6th ed. St. Louis, MO: Mosby, 1980:17–20, with permission.) |
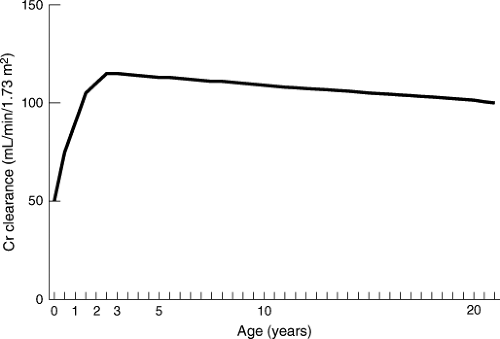 Figure 3.5. Change in endogenous creatinine (Cr) clearance during childhood. (Adapted from Habersang RWO. Dosage. In: Shirkey HC, ed. Pediatric therapy, 6th ed. St. Louis, MO: Mosby, 1980:17–20, with permission.) |
Influence of Development on Drug Biodisposition and Action
Drug Absorption
Developmental changes in the GI tract are important because medications are commonly administered to children by mouth, and maturational changes may influence drug absorption. It is difficult to generalize, however, because differences in absorption of orally administered drugs associated with growth and maturation are not readily predicted from known drug or developmental characteristics. Nevertheless, it is important to be aware of those aspects of GI tract development that may influence drug absorption.
Most drugs are absorbed from the gut by passive diffusion across a concentration gradient, although active transport mechanisms may be involved for a minority of drugs. The rate and extent of absorption are influenced by physical–chemical properties of the drug and characteristics of the recipient’s gut. Important drug properties include molecular weight, lipid solubility, pKa (dissociation constant of weak acids or bases), formulation in which the drug is administered (e.g., liquid, solid, extended release), and disintegration time and dissolution rate of solid dose forms. Important recipient characteristics include gastric acidity, gastric emptying, gut motility, gut surface area, gut drug-metabolizing enzymes and active transporters, secretion of bile acids and pancreatic lipases, first-pass metabolism, diet at different ages, and diurnal variation.
Reflux of gastric contents retrograde into the esophagus is very common during the first year of life (19). Excessive gastroesophageal reflux may result in regurgitation of medication, resulting in variable and unpredictable loss of an orally administered dose.
Gastric emptying is an important determinant of rate of absorption because most drug absorption takes place in the duodenum. Delayed gastric emptying in infants not only contributes to gastroesophageal reflux but also may result in delayed drug absorption (20,21,22). On the other hand, gastric emptying in prepubescent children is equal to or exceeds that in adults. This tends to facilitate more rapid drug absorption, other factors being equal (21,23). Administration of medication in liquid as opposed to solid dose forms, as is commonly the case for children, also increases the rate of absorption. In contrast, shorter GI transit time in young children may actually reduce the fraction of dose absorbed when drugs are administered in sustained-release formulations (24,25).
Decreased gastric acid production in the younger infant may result in increased bioavailability of acid-labile drugs such as the penicillins (22,26). For example, increased absorption of penicillin G, ampicillin, and nafcillin in infants compared with older children and adults has been reported (20). However, perturbation of drug absorption due to reduction in gastric acidity is negligible beyond infancy.
Rate and extent of drug absorption are determined to a significant degree by the absorptive surface area of the duodenum. Greater relative small gut surface area in young children tends to enhance drug absorption.
Drugs absorbed from the intestine into the portal circulation are delivered to the liver before entering the systemic circulation. High hepatic extraction of some drugs on the first pass through the liver results in removal of a large fraction of the absorbed drug by the liver, resulting in decreased systemic bioavailability. Intestinal metabolism of drugs also may decrease systemic bioavailability. Little is known about the effect of intestinal and hepatic maturation on first-pass uptake of drugs. However, one would predict, based on increased hepatic clearance in children, that first-pass processes in children beyond infancy would be equal to or exceed uptake in adults. Wilson et al. (27) described wide intersubject variability and low serum concentrations of two high-uptake drugs, propoxyphene and propanolol, when administered orally to children 2 to 13 years of age. This is consistent with extensive first-pass uptake.
Maturation of gut flora during childhood modifies digoxin metabolism. Reduction of digoxin to inactive metabolites by anaerobic GI bacteria accounts for a significant fraction of digoxin clearance in approximately 10% of adult patients (28). Reduction metabolites are not detected in children until after 16 months of age, and the adult metabolite pattern is not found until 9 years of age (29).
The rectum is an alternative route of enteral drug administration in children, which may be used when vomiting or other intervening conditions preclude oral dosing. Drugs administered rectally are absorbed into the hemorrhoidal veins, which are part of the systemic rather than the portal circulation. First-pass uptake, therefore, is not a consideration with rectal administration. However, rectal dosing is less than satisfactory in many cases for other reasons. Absorption of drugs administered in suppository form is typically erratic and incomplete. Furthermore, presence of feces in the rectal vault impedes absorption. In younger children and infants, the dose may be expelled before absorption is complete, thereby reducing bioavailability to a variable extent. Nevertheless, some medications may be successfully administered rectally in solution (30). These include diazepam and valproic acid for seizures and phenobarbital for seizures, sedation, or preanesthesia (31,32). In addition, rectal corticosteroids are routinely used in the treatment of inflammatory bowel disease.
Absorption of drugs from intramuscular or subcutaneous injection sites is influenced by characteristics of the patient as well as properties of the injected drug. Blood flow to the injection site, muscle mass, quantity of adipose tissue, and muscle activity are patient characteristics that determine the rate and extent of absorption. Solubility of the drug at the pH of extracellular fluid, ease with which the drug diffuses across capillary membranes, and surface area over which the injection volume spreads also determine absorption (21,22,30,31,33). Extravascular injection is not an optimal route of administration in the presence of hypoperfusion syndromes, dehydration, vasomotor instability, starvation, or cachexia because all these conditions impede absorption. Some drugs, such as erythromycin and certain cephalosporin antibiotics, are not usually administered intramuscularly because they cause unacceptable pain and tissue trauma. However, many drugs, including most aminoglycoside and penicillin antibiotics, may be administered intramuscularly with resulting plasma concentrations comparable with those achieved with intravenous
administration. Conversely, highly hydrophobic drugs such as diazepam and phenytoin are not absorbed well following intramuscular injection and should not be given by this route. Furthermore, phenytoin forms insoluble crystals in intramuscular injection sites, associated with local hemorrhage, muscle necrosis, and minimal systemic absorption (34). Fosphenytoin, which is water soluble, is preferable to phenytoin for parenteral administration.
administration. Conversely, highly hydrophobic drugs such as diazepam and phenytoin are not absorbed well following intramuscular injection and should not be given by this route. Furthermore, phenytoin forms insoluble crystals in intramuscular injection sites, associated with local hemorrhage, muscle necrosis, and minimal systemic absorption (34). Fosphenytoin, which is water soluble, is preferable to phenytoin for parenteral administration.
Drug Distribution
The distribution of a drug throughout the body is influenced by the binding affinity of the drug for plasma and tissue proteins, the lipid/water solubility partition of the drug, the molecular weight of the drug, and the degree of ionization of the drug at physiologic pH. Age-dependent changes in body composition (see earlier in text) may influence drug distribution in the developing child. Highly lipid-soluble compounds such as inhalation anesthetics and lipophilic sedative/hypnotic agents typically exhibit relatively larger distribution volumes in infants during the first year of life compared with older children because of the relatively larger proportion of body fat in infants. Likewise, the apparent distribution volume of drugs such as penicillin, aminoglycoside, and cephalosporin antibiotics, which are distributed primarily in extracellular water, tends to be greater in infants and decreases during maturation coincident with the progressive relative decrease in extracellular water (21,23,33). In general, the apparent volume of distribution of drugs tends to be greater in infants and decreases toward adult values during childhood. However, there is a great deal of interindividual variation, and important exceptions to this general rule exist. Examples of such exceptions are theophylline (35) and phenobarbital (36), which show little consistent age-related change in distribution volume.
Although the plasma protein binding of many drugs is decreased in the fetus and newborn infant relative to adults, age-related differences in plasma protein binding are not clinically significant beyond the newborn period (37). However, maturational changes in tissue binding can significantly affect drug distribution. For example, the myocardial-to-plasma digoxin concentration in infants and children up to 36 months of age is two to three times that of adults (38,39,40). Using specific assay methods, increased myocardial digoxin concentrations relative to adults have been demonstrated in children and these do not appear to be due to assay interference by endogenous digoxin-like substances (41). In addition, erythrocytes from infants bind three times the quantity of digoxin as adult erythrocytes (40). The increased myocardial and erythrocyte binding of digoxin is associated with a significantly greater volume of distribution of digoxin in infants and children compared with adults.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


