Chapter 604 Disorders of Neuromuscular Transmission and of Motor Neurons
604.1 Myasthenia Gravis
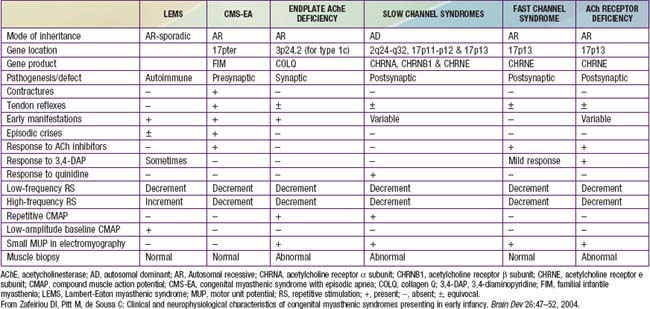
Myasthenia gravis is a chronic disease characterized by rapid fatigability of striated muscle. The most common cause is an immune-mediated neuromuscular blockade. The release of acetylcholine (ACh) into the synaptic cleft by the axonal terminal is normal, but the postsynaptic muscle membrane or motor endplate is less responsive than normal. A decreased number of available ACh receptors is due to circulating receptor-binding antibodies in most cases of acquired myasthenia. The disease is generally not hereditary and is an autoimmune disorder. A rare familial myasthenia gravis is probably an autosomal recessive trait and is not associated with plasma anti-ACh antibodies. One familial form is a deficiency of motor endplate acetylcholinesterase (AChE). Infants born to myasthenic mothers can have a transient neonatal myasthenic syndrome secondary to placentally transferred anti-ACh receptor antibodies, distinct from congenital myasthenia gravis (Table 604-1).
Table 604-1 CLINICAL, PATHOLOGIC, AND NEUROPHYSIOLOGIC CHARACTERISTICS OF VARIOUS CONGENITAL MYASTHENIC SYNDROMES
Clinical Manifestations
The syndrome of transient neonatal myasthenia gravis is to be distinguished from a rare and often hereditary congenital myasthenia gravis not related to maternal myasthenia that is nearly always a permanent disorder without spontaneous remission (see Table 604-1). Several distinct genetic forms are recognized, all with onset at birth or in early infancy with hypotonia, ophthalmoplegia, ptosis, dysphagia, weak cry, facial weakness, easy muscle fatigue generally, and sometimes respiratory insufficiency or failure, the last often precipitated by a minor respiratory infection. Cholinesterase inhibitors have a favorable effect in most, but in some forms the symptoms and signs are actually worsened. Most congenital myasthenic syndromes are transmitted as autosomal recessive traits, but the slow channel syndrome is autosomal dominant. Five defective postsynaptic molecules have been identified in the pathogenesis of congenital myasthenia gravis and account for 85% of cases; rapsyn may be the most common. Acetylcholine receptor deficiencies have >60 identified genetic mutations. Anti-AChR and anti-MuSK antibodies are absent in serum, unlike autoimmune forms of myasthenia gravis affecting older children and adults.
Laboratory Findings and Diagnosis
Recommendations on the Use of Cholinesterase Inhibitors as a Diagnostic Test for Myasthenia Gravis in Infants and Children
Children 2 Years and Older
For Children Younger than 2 Years
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


