Chapter 330 Disorders of Malabsorption
All disorders of malabsorption are associated with diminished intestinal absorption of one or more dietary nutrients. Malabsorption can result from a defect in the nutrient digestion in the intestinal lumen or from defective mucosal absorption. Malabsorption disorders can be categorized into generalized mucosal abnormalities usually resulting in malabsorption of multiple nutrients (Table 330-1) or malabsorption of specific nutrients (carbohydrate, fat, protein, vitamins, minerals, and trace elements) (Table 330-2). Almost all the malabsorption disorders are accompanied by chronic diarrhea (Chapter 333).
Table 330-1 MALABSORPTION DISORDERS AND CHRONIC DIARRHEA ASSOCIATED WITH GENERALIZED MUCOSAL DEFECT
IgA, immunoglobulin A.
Table 330-2 CLASSIFICATION OF MALABSORPTION DISORDERS AND CHRONIC DIARRHEA BASED ON THE PREDOMINANT NUTRIENT MALABSORBED
CARBOHYDRATE MALABSORPTION
FAT MALABSORPTION
AMINO ACID MALABSORPTION
MINERAL AND VITAMIN MALABSORPTION
DRUG INDUCED
Clinical Approach
The clinical features depend on the extent and type of the malabsorbed nutrient. The common presenting features, especially in toddlers with malabsorption, are diarrhea, abdominal distention, and failure to gain weight, with a fall in growth chart percentiles. Physical findings include muscle wasting and the disappearance of the subcutaneous fat, with subsequent loose skinfolds (Fig. 330-1). The nutritional consequences of malabsorption are more dramatic in toddlers because of the limited energy reserves and higher proportion of calorie intake being used for weight gain and linear growth. In older children, malnutrition can result in growth retardation, as is commonly seen in children with late diagnosis of celiac disease. If malabsorption is untreated, linear growth slows, and with prolonged malnutrition, death can follow (Chapter 43). This extreme outcome is usually restricted to children living in the developing world, where resources to provide enteral and parenteral nutrition support may be limited. Specific findings on examination can guide toward a specific disorder; edema is usually associated with protein-losing enteropathy, digital clubbing with cystic fibrosis and celiac disease, perianal excoriation and gaseous abdominal distention with carbohydrate malabsorption, perianal and circumoral rash with acrodermatitis enteropathica, abnormal hair with Menkes syndrome, and the typical facial features diagnostic of the Johanson-Blizzard syndrome.
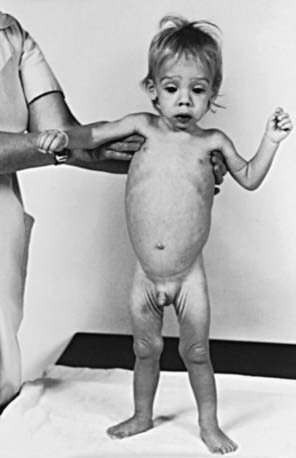
The nutritional assessment is an important part of clinical evaluation in children with malabsorptive disorders (Chapter 41). Long-term calcium and vitamin D malabsorption can lead to reduced bone mineral density and metabolic bone disease, with increased risk of bone fractures. Vitamin K malabsorption, irrespective of the underlying mechanism (fat malabsorption, mucosal atrophy), can result in coagulopathy. Severe protein-losing enteropathy is often associated with malabsorption syndromes (celiac disease, intestinal lymphangiectasia) and causes hypoalbuminemia and edema. Other nutrient deficiencies include iron malabsorption causing microcytic anemia and low reticulocyte count, low serum folate levels in conditions associated with mucosal atrophy, and low serum vitamin A and vitamin E concentrations in fat malabsorption.
Clinical history alone might not be sufficient to make a specific diagnosis, but it can direct the pediatrician toward a more structured and rational investigative approach. Diarrhea is the main clinical expression of malabsorption. Onset of diarrhea in early infancy suggests a congenital defect (Table 330-3). In secretory diarrhea due to disorders such as congenital chloride diarrhea and microvillus inclusion disease, the stool is watery and voluminous and can be mistaken for urine. Onset of symptoms after introduction of a particular food into a child’s diet can provide diagnostic clues, such as with sucrose in sucrase-isomaltase deficiency. The nature of the diarrhea may be helpful: explosive watery diarrhea suggests carbohydrate malabsorption; loose, bulky stools are associated with celiac disease; and pasty and yellowish offensive stools suggest an exocrine pancreatic insufficiency. Stool color is usually not helpful; green stool with undigested “peas and carrots” can suggest rapid intestinal transit in toddler’s diarrhea, which is a self-limiting condition unassociated with failure to thrive.
Table 330-3 DIARRHEAL DISEASES APPEARING IN THE NEONATAL PERIOD
| CONDITION | CLINICAL FEATURES |
|---|---|
| Microvillus inclusion disease | Secretory watery diarrhea |
| Tufting enteropathy | Secretory watery diarrhea |
| Congenital glucose-galactose malabsorption | Acidic diarrhea |
| Congenital lactase deficiency | Acidic diarrhea |
| Congenital chloride diarrhea | Hydramnion, secretory watery diarrhea Metabolic alkalosis |
| Congenital defective jejunal Na+/H+ exchange | Hydramnion, secretory watery diarrhea |
| Congenital bile acid malabsorption | Steatorrhea |
| Congenital enterokinase deficiency | Failure to thrive, edema |
| Congenital trypsinogen deficiency | Failure to thrive, edema |
| Congenital lipase and/or co-lipase deficiency | Failure to thrive, oily stool |
| Enteric anendocrinosis (NEUROG 3 mutation) | Hyperchloremic acidosis, failure to thrive |
Adapted from Schmitz J: Maldigestion and malabsorption. In Walker WA, Durie PR, Hamilton JR, et al, editors: Pediatric gastrointestinal disease, ed 3, Hamilton, Ontario, 2000, BC Decker, p 55.
330.1 Evaluation of Children with Suspected Intestinal Malabsorption
Michael J. Lentze and David Branski
Investigations for Fat Malabsorption
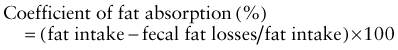
where fat intake and fat losses are in grams. Because fecal fat balance studies are cumbersome, expensive, and unpleasant to perform, simpler tests are often preferred. Among these stool tests, the acid steatocrit test is the most reliable. When bile acid deficiency is suspected of being the cause of fat malabsorption, the evaluation of bile acid levels in duodenal fluid aspirate may be useful.
Investigations for Exocrine Pancreatic Function (Fig. 330-2)
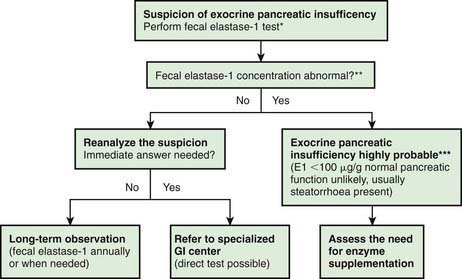
The gold standard test for exocrine pancreatic function is direct analysis of duodenal aspirate for volume, bicarbonate, trypsin and lipase upon secretin and pancreozymin/cholecysto-kinin stimulation. This involves duodenal intubation, and only a few centers perform this test (Chapter 340).
330.2 Gluten-Sensitive Enteropathy (Celiac Disease)
Etiology and Epidemiology
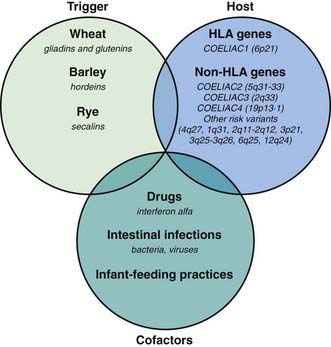
Celiac disease is a common disorder (1% prevalence of biopsy-proven disease). It is thought to be rare in Central Africa and East Asia. Environmental factors might affect the risk of developing celiac disease or the timing of its presentation. Prolonged breastfeeding has been associated with a reduced incidence of symptomatic disease. Less clear is the effect of the time of gluten introduction in the infant diet; the ingestion of increased amounts of gluten in the 1st year of life can increase the incidence. Infectious agents have been hypothesized to play a role because frequent rotavirus infections are associated with an increased risk. It is plausible that the contact with gliadin at a time when there is ongoing intestinal inflammation, altered intestinal permeability, and enhanced antigen presentation can increase the risk of developing the disease, at least in a subset of persons (Fig. 330-3).
Clinical Presentation and Associated Disorders
Clinical features of celiac disease vary considerably (Table 330-4). Intestinal symptoms are common in children whose disease is diagnosed within the 1st 2 years of life; failure to thrive, chronic diarrhea, vomiting, abdominal distention, muscle wasting, anorexia, and irritability are present in most cases (see Fig. 330-1). Occasionally there is constipation, rectal prolapse, or intussusception. As the age at presentation of the disease shifts to later in childhood, and with the more liberal use of serologic screening tests, extraintestinal manifestations and associated disorders, without any accompanying digestive symptoms, have increasingly become recognized, affecting almost all organs (Table 330-5).
Table 330-4 SOME CLINICAL MANIFESTATIONS OF CELIAC DISEASE IN CHILDREN AND ADOLESCENTS
| SYSTEM | MANIFESTATION | (POSSIBLE) CAUSE |
|---|---|---|
| Gastrointestinal | Diarrhea Distended abdomen Vomiting Anorexia Weight loss Failure to thrive Aphthous stomatitis | Atrophy of the small bowel mucosa Malabsorption |
| Hematologic | Anemia | Iron malabsorption |
| Skeletal | Rickets Osteoporosis Enamel hypoplasia of the teeth | Calcium/vitamin D malabsorption |
| Muscular | Atrophy | Malnutrition |
| Neurologic | Peripheral neuropathy Epilepsy Irritability | Thiamine/vitamin B12 deficiency |
| Endocrinologic | Short stature Pubertas tarda Secondary hyperparathyroidism | Malnutrition Calcium/vitamin D malabsorption |
| Dermatologic | Dermatitis herpetiformis Alopecia areata Erythema nodosum | Autoimmunity |
| Respiratory | Idiopathic pulmonary hemosiderosis |
Adapted from Mearin ML: Celiac disease among children and adolescents, Curr Prob Pediatr Adolesc Health Care 37:81–112, 2007.
Table 330-5 RISK GROUPS FOR CELIAC DISEASE CASE-FINDING
IgA, immunoglobulin A.
Modified from Di Sabatino A, Corazza GR: Coeliac disease, Lancet 373:1480–1490, 2009.
Silent celiac disease is being increasingly recognized, mainly in asymptomatic 1st-degree relatives of celiac patients investigated during screening studies. However, small bowel biopsy in these people reveals severe mucosal damage consistent with celiac disease. Potential celiac disease is defined when patients are identified by positive screening studies but without documented celiac disease on small bowel biopsy. It is important to follow these patients because they can develop established celiac disease in the future (Table 330-6).
Table 330-6 CLINICAL SPECTRUM OF CELIAC DISEASE
SYMPTOMATIC
Frank malabsorption symptoms: chronic diarrhea, failure to thrive, weight loss
Extraintestinal manifestations: anemia, fatigue, hypertransaminasemia, neurologic disorders, short stature, dental enamel defects, arthralgia, aphthous stomatitis
SILENT
No apparent symptoms in spite of histologic evidence of villous atrophy
In most cases identified by serologic screening in at-risk groups (see Table 330-1)
LATENT
Subjects who have a normal histology, but at some other time, before or after, have shown a gluten-dependent enteropathy
POTENTIAL
Subjects with positive celiac disease serology but without evidence of altered jejunal histology
It might or might not be symptomatic
Diagnosis
The ultimate diagnosis of celiac disease relies on the demonstration of specific, though not pathognomonic, histopathologic abnormalities in the small bowel mucosa (Table 330-7). According to The European Society for Pediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) current criteria, the 2 requirements mandatory for the diagnosis of celiac disease are the finding of villous atrophy with hyperplasia of the crypts and abnormal surface epithelium, while the patient is eating adequate amounts of gluten, and a full clinical remission after withdrawal of gluten from the diet. The finding of circulating IgA celiac disease–associated antibodies at the time of diagnosis and their disappearance on a gluten-free diet adds weight to the diagnosis. A control biopsy to verify the consequences of the gluten-free diet on the mucosal architecture is considered mandatory only in patients with an equivocal clinical response to the diet. Gluten challenge is not considered mandatory except in situations where there is doubt about the initial diagnosis, for example, when an initial biopsy was not performed or when the biopsy specimen was inadequate or atypical of celiac disease.
Table 330-7 OTHER CAUSES OF FLAT MUCOSA
Modified from Di Sabatino A, Corazza GR: Coeliac disease, Lancet 373:1480–1490, 2009.
Treatment
The only treatment for celiac disease is lifelong strict adherence to a gluten-free diet (Fig. 330-4). This requires a wheat-, barley-, and rye-free diet. Despite evidence that oats are safe for most patients with celiac disease, there is concern regarding the possibility of contamination of oats with gluten during harvesting, milling, and shipping. Nevertheless, it seems wise to add oats to the gluten-free diet only when the latter is well established, so that possible adverse reactions can be readily identified. There is a consensus that all celiac disease patients should be treated with a gluten-free diet regardless of the presence of symptoms. However, whereas it is relatively easy to assess the health improvement after treatment of celiac disease in patients with clinical symptoms of the disease, it proves difficult in persons with asymptomatic celiac disease. The nutritional risks, particularly osteopenia, are those mainly feared for subjects who have silent celiac disease and continue on a gluten-containing diet. Little is known about the health risks in untreated patients with minor enteropathy, which may be clinically silent. There are no guidelines concerning the need for a gluten-free diet in subjects with “potential” celiac disease (patients with positive celiac disease–associated serology but without enteropathy).
Branski D, Fasano A, Troncone R. Latest development in the pathogenesis and treatment of celiac disease. J Pediatr. 2006;149:295-300.
Branski D, Troncone R. Celiac disease: a reappraisal. J Pediatr. 1998;133:181-187.
Collin P, Huhtala H, Virta L, et al. Diagnosis of celiac disease in clinical practice: physician’s alertness to the condition essential. J Clin Gastroenterol. 2007;41:152-156.
Di Sabatino A, Corazza RG. Coeliac disease. Lancet. 2009;373:1480-1493.
Dubois PC, Van Heel DA. Translational mini-review series on the immunogenetics of gut disease: immunogenetics of celiac disease. Clin Exp Immunol. 2008;153:162-173.
Ford AC, Chey WD, Talley NJ, et al. Yield of diagnostic tests for celiac disease in individuals with symptoms suggestive of irritable bowel syndrome. Arch Intern Med. 2009;169:651-658.
Green PHR, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731-1743.
Jones R, Sleet S. Coeliac disease. BMJ. 2009;338:539-540.
Kline RM, Neudorf SML, Baron HI. Correction of celiac disease after allogeneic hematopoietic stem cell transplantation for acute myelogenous leukemia. Pediatrics. 2007;120:e1120-e1122.
Kurppa K, Ashorn M, Iltanen S, et al. Celiac disease without villous atrophy in children: a prospective study. J Pediatr. 2010;157:373-380.
Ludvigsson JF, Montgomery SM, Ekbom A, et al. Small-intestinal histopathology and mortality risk in celiac disease. JAMA. 2009;302:1171-1178.
McGowan KE, Castiglione DA, Butzner JD. The changing face of childhood celiac disease in North America: Impact of serological testing. Pediatrics. 2009;124:1572-1578.
Mearin ML. Celiac disease among children and adolescents. Curr Prob Pediatr Adolesc Health Care. 2007;37:81-112.
Meresse B, Ripoche J, Heyman M, et al. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009;2:8-23.
Olsson C, Hernell O, Hornell A, et al. Difference in celiac disease risk between Swedish birth cohorts suggests an opportunity for primary prevention. Pediatrics. 2008;122:528-534.
Rashtak S, Murray JA. Tailored testing for celiac disease. Ann Intern Med. 2007;147:339-340.
Richey R, Howdle P, Shaw E, et al. Recognition and assessment of coeliac disease in children and adults: summary of NICE guidance. BMJ. 2009;338:1386-1388.
Rodrigues AF, Jenkins HR. Investigation and management of coeliac disease. Arch Dis Child. 2008;93:251-254.
Sattar N, Lazare F, Kacer M, et al. Celiac disease in children, adolescents, and young adults with autoimmune disease. J Pediatr. 2011;158:272-275.
Simmons JH, Klingensmith GJ, McFann K, et al. Celiac autoimmunity in children with type 1 diabetes: a two-year follow-up. J Pediatr. 2011;158:276-281.
Smyth DJ, Plagnol V, Walker NM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767-2776.
Sollid LM, Lundin KEA. Diagnosis and treatment of celiac disease. Mucosal Immunol. 2009;2:3-7.
Telega G, Bennet TR, Welin S. Emerging new clinical patterns in the presentation of celiac disease. Arch Pediatr Adolesc Med. 2008;162:164-168.
Troncone R, Ivarsson A, Szajewska H, et al. Review article: future research on celiac disease—a position report from the European multistakeholder platform on celiac disease (CDEUSSA). Aliment Pharmacol Ther. 2008;27:1030-1043.
van Koppen EJ, Schweizer JJ, Csizmadia CGDS, et al. Long-tern health and quality-of-life consequences of mass screening for childhood celiac disease: a 10-year follow-up study. Pediatrics. 2009;123:e582-e588.
Zanchi C, Di Leo G, Ronfani L, et al. Bone metabolism in celiac disease. J Pediatr. 2008;153:262-265.
330.3 Other Malabsorptive Syndromes
Congenital Intestinal Mucosal Defects
Microvillus Inclusion Disease (Congenital Microvillus Atrophy)
Microvillus inclusion disease is an autosomal recessive disorder, which manifests at birth with profuse watery secretory diarrhea. It is the most commonly recognized cause of congenital diarrhea. Light microscopy of the small bowel mucosa demonstrates diffuse thinning of the mucosa, with hypoplastic villus atrophy and no inflammatory infiltrate. Diagnosis may be easily performed with light microscopy using PAS and CD10 staining, which shows a very thin or absent brush border, together with positive PAS and CD10 intracellular inclusions. Electron microscopy shows enterocytes with absent or sparse microvilli. The apical cytoplasm of the enterocytes contains electron-dense secretory granules; the hallmark is presence of microvilli within involutions of the apical membrane. Polyhydramnios is not a classic presentation of MID. Neonates usually present with dehydration and failure to thrive. Despite parenteral nutrition, diarrhea continues and initial fluid management is difficult. The disease is fatal without long-term parenteral nutrition support. Some infants present with rapid onset of liver disease, which is associated with pruritus. Most children die in infancy or early childhood. The long-acting somatostatin analog octreotide has been used as treatment and can reduce the volume of stool in some infants (Chapter 331). Intestinal transplantation is the only definitive treatment for this rare disease. Rarely, in milder forms of the disease, the patient can reach young adulthood and enjoy partially oral feeding. The underlying gene defect is a mutation in MYO5B, which encodes a protein involved in subcellular protein trafficking. Several types of mutations are involved.



