Chapter 650 Disorders of Keratinization
Disorders of Cornification
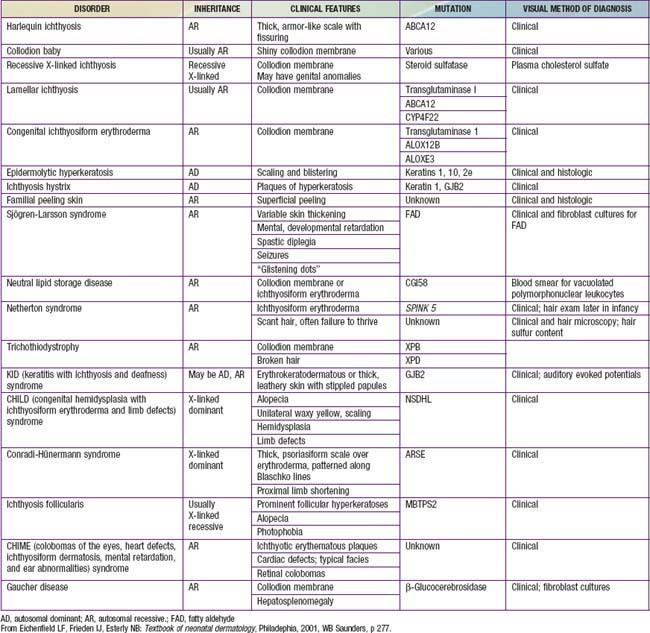
Disorders of cornification (ichthyoses) are a primary group of inherited conditions characterized clinically by patterns of scaling and histopathologically by hyperkeratosis. They are usually distinguishable on the basis of inheritance patterns, clinical features, associated defects, and histopathologic changes (Table 650-1).
Collodion Baby
Clinical Manifestations
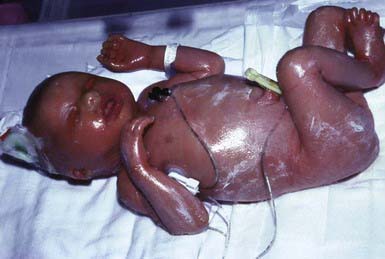
Collodion babies are covered at birth by a thick, taut membrane resembling oiled parchment or collodion (Fig. 650-1), which is subsequently shed. Affected neonates have ectropion, flattening of the ears and nose, and fixation of the lips in an O-shaped configuration. Hair may be absent or may perforate the abnormal covering. The membrane cracks with initial respiratory efforts and, shortly after birth, begins to desquamate in large sheets. Complete shedding may take several weeks, and a new membrane may occasionally form in localized areas.
Lamellar Ichthyosis and Congenital Ichthyosiform Erythroderma (Nonbullous Congenital Ichthyosiform Erythroderma)
Clinical Manifestations
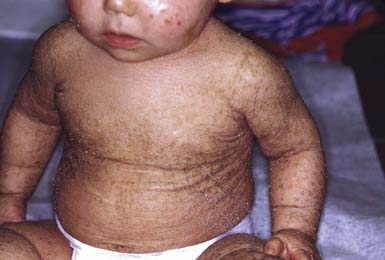
After shedding of the collodion membrane, if present, lamellar ichthyosis evolves into large, quadrilateral, dark scales that are free at the edges and adherent at the center. Scaling is often pronounced and involves the entire body surface, including flexural surfaces (Fig. 650-2). The face is often markedly involved, including ectropion and small, crumpled ears. The palms and soles are generally hyperkeratotic. The hair may be sparse and fine, but the teeth and mucosal surfaces are normal. Unlike in congenital ichthyosiform erythroderma, there is little erythema.
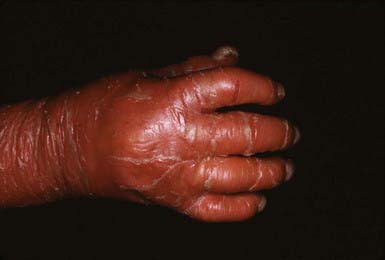
In congenital ichthyosiform erythroderma, erythroderma tends to be persistent, and scales, although they are generalized, are finer and whiter than in lamellar ichthyosis (Fig. 650-3). Hyperkeratosis is particularly noticeable around the knees, elbows, and ankles. Palms and soles are uniformly hyperkeratotic. Patients have sparse hair, cicatricial alopecia, and nail dystrophy. Neither form includes blistering.
Ichthyosis Vulgaris
Clinical Manifestations
Ichthyosis vulgaris is the most common of the disorders of keratinization, with an incidence of 1/250 live births. Onset generally occurs in the 1st yr of life. In most cases, it is trivial, consisting only of slight roughening of the skin surface. Scaling is most prominent on the extensor aspects of the extremities, particularly the legs (Fig. 650-4). Flexural surfaces are spared, and the abdomen, neck, and face are relatively uninvolved. Keratosis pilaris, particularly on the upper arms and thighs, accentuated markings, and hyperkeratosis on the palms and soles, and atopy are relatively common. Scaling is most pronounced during the winter months and may abate completely during warm weather. There is no accompanying disorder of hair, teeth, mucosal surfaces, or other organ systems.