Chapter 433 Diseases of the Myocardium
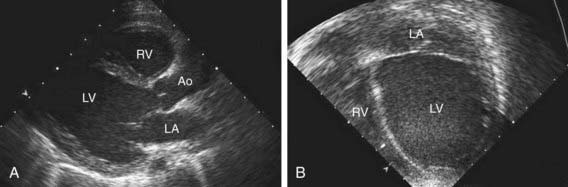
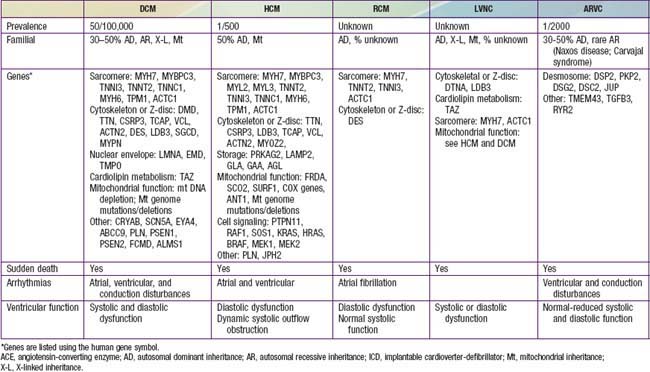
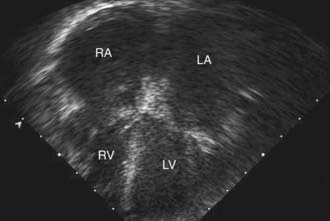
Table 433-1 classifies the cardiomyopathies based on their anatomic (ventricular morphology) and functional pathophysiology. Dilated cardiomyopathy, the most common form of cardiomyopathy, is characterized predominantly by left ventricular dilation and decreased left ventricular systolic function (Fig. 433-1). Hypertrophic cardiomyopathy demonstrates increased ventricular myocardial wall thickness, normal or increased systolic function, and often, diastolic (relaxation) abnormalities (Table 433-2). Restrictive cardiomyopathy is characterized by nearly normal ventricular chamber size and wall thickness with preserved systolic function, but dramatically impaired diastolic function leading to elevated filling pressures and atrial enlargement (see Fig. 433-3). Arrhythmogenic right ventricular cardiomyopathy and left ventricular non-compaction are characterized by specific morphologic abnormalities and heterogeneous functional disturbances.
Table 433-1 ETIOLOGY OF PEDIATRIC MYOCARDIAL DISEASE
| CARDIOMYOPATHY | |
| Dilated Cardiomyopathy (DCM) | |
| Neuromuscular diseases | Muscular dystrophies (Duchenne, Becker, limb girdle, Emery- Dreifuss, congenital muscular dystrophy, etc.), myotonic dystrophy, myofibrillar myopathy |
| Inborn errors of metabolism | Fatty acid oxidation disorders (trifunctional protein, VLCAD), carnitine abnormalities (carnitine transport, CPTI, CPTII), mitochondrial disorders (including Kearns-Sayre syndrome), organic acidemias (propionic acidemia) |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic DCM |
| Genetic syndromes | Alstrom syndrome, Barth syndrome (phospholipid disorders) |
| Ischemic | Most common in adults |
| Chronic tachyarrhythmias | |
| Hypertrophic Cardiomyopathy (HCM) | |
| Inborn errors of metabolism | Mitochondrial disorders (including Friedreich ataxia, mutations in nuclear or mitochondrial genome), storage disorders (glycogen storage disorders, especially Pompe; mucopolysaccharidoses; Fabry disease; sphingolipidoses; hemochromatosis; Danon disease) |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic HCM |
| Genetic syndromes | Noonan, Costello, cardio-faciocutaneous, Beckwith-Wiedemann syndrome |
| Infant of a diabetic mother | Transient hypertrophy |
| Restrictive Cardiomyopathy (RCM) | |
| Neuromuscular disease | Myofibrillar myopathies |
| Metabolic | Storage disorders |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic RCM |
| Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) | |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic ARVC |
| Left Ventricular Noncompaction (LVNC) | X-linked (Barth syndrome), autosomal dominant, autosomal recessive, mitochondrial inheritance, or sporadic LVNC Sporadic LVNC |
| SECONDARY OR ACQUIRED MYOCARDIAL DISEASE | |
| Myocarditis | Viral: parvovirus B19, adenovirus, coxsackievirus A and B, echovirus, rubella, varicella, influenza, mumps, Epstein-Barr virus, cytomegalovirus, measles, poliomyelitis, smallpox vaccine, hepatitis C virus, HIV virus, or opportunistic infections Bacterial: diphtheria, mycoplasma, meningococcus, leptospirosis, Lyme disease, typhoid fever, tuberculosis, streptococcus, listeriosis Parasitic: Chagas disease, toxoplasmosis, Loa loa, Toxocara canis, schistosomiasis, cysticercosis, echinococcus, trichinosis |
| Systemic Inflammatory Disease | Systemic lupus erythematosus (SLE), infant of mother with SLE, scleroderma, Churg-Strauss vasculitis, rheumatoid arthritis, rheumatic fever, sarcoidosis, dermatomyositis, periarteritis nodosa, hypereosinophilic syndrome (Löffler syndrome), acute eosinophilic necrotizing myocarditis, giant cell myocarditis |
| Nutritional Deficiency | Beriberi (thiamine deficiency), kwashiorkor, Keshan disease (selenium deficiency) |
| Drugs, Toxins | Doxorubicin (Adriamycin), cyclophosphamide, chloroquine, ipecac (emetine), sulfonamides, mesalazine, chloramphenicol, alcohol, hypersensitivity reaction, envenomations, irradiation, herbal remedy (blue cohosh) |
| Coronary Artery Disease | Kawasaki disease, medial necrosis, anomalous left coronary artery from the pulmonary artery (ALCAPA), other congenital coronary anomalies (anomalous right coronary, coronary ostial stenosis), familial hypercholesterolemia |
| Hematology-Oncology | Anemia, sickle cell disease, leukemia |
| Endocrine-Neuroendocrine | Hyperthyroidism, carcinoid tumor, pheochromocytoma |
Bibliography
Judge DP: Use of genetics in the clinical evaluation of cardiomyopathy, JAMA 302:2471–2476, 2009.
433.1 Dilated Cardiomyopathy
Etiology and Epidemiology
Dilated cardiomyopathy (DCM), the most common form of cardiomyopathy in children, is the cause of significant morbidity and mortality as well as a common indication for cardiac transplantation. The etiologies are diverse. Unlike adult patients with dilated cardiomyopathy, ischemic etiologies are rare in children, although these include anomalous origin of the left coronary artery from the pulmonary artery, premature coronary atherosclerosis (homozygous type II hypercholesterolemia), and coronary inflammatory diseases, such as Kawasaki disease. It is estimated that up to 50% of cases are genetic, including some with metabolic causes (see Table 433-1). Although the most common etiology of dilated cardiomyopathy remains idiopathic, it is likely that undiagnosed familial/genetic conditions and myocarditis predominate. The annual incidence of DCM in children younger than 18 yr is 0.57 cases per 100,000 per year. Incidence is higher in males, African Americans, and in infants less than 1 yr old.
Pathogenesis
In 20-50% of cases, the DCM is familial with autosomal dominant inheritance most common (see Table 433-2). Duchenne and Becker muscular dystrophies (Chapter 601.1) are X-linked cardiomyopathies that account for 5-10% of familial dilated cardiomyopathy cases. These dystrophinopathies result in an abnormal sarcomere-cytoskeleton connection, causing impaired myocardial force generation, myocyte damage/scarring, chamber enlargement, and altered function.
Laboratory Findings
Electrocardiographic screening reveals atrial or ventricular hypertrophy, nonspecific T-wave abnormalities, and occasionally atrial or ventricular arrhythmias. The chest x-ray demonstrates cardiomegaly and may reveal pulmonary vascular prominence or pleural effusions. The echocardiogram is often diagnostic, demonstrating the characteristic findings of left ventricular enlargement, decreased ventricular contractility, and occasionally a globular (remodeled) left ventricular contour (see Fig. 433-1). Right ventricular enlargement and depressed function are occasionally noted. Echo Doppler studies can reveal evidence of pulmonary hypertension, mitral regurgitation, or other structural cardiac or coronary abnormalities.
Additional testing should include CBC, renal and liver function tests, CPK, cardiac troponin I, lactate, plasma amino acids, urine organic acids, and an acylcarnitine profile. Additional genetic and enzymatic testing may be useful (see Table 433-2). Cardiac catheterization and endomyocardial biopsy are not routine but may be useful in patients with acute dilated cardiomyopathy. Biopsy samples can also be assessed for the presence of mononuclear cell infiltrates, myocardial damage, storage abnormalities, and viral infection or genomes. It is important to consider screening of 1st-degree family members utilizing echocardiography and ECG.



