Most cancers arise due to accumulation of genetic alterations in several genes involved in cell growth and DNA repair. In contrast to “sporadic” cancers, in which all mutations are acquired, hereditary cancers arise in individuals who have inherited a mutation in a cancer-causing gene. These individuals generally develop cancer at a younger age than sporadic cases, may develop multiple tumors in a single organ, or have bilateral development of tumors in paired organs and they often develop multiple primary cancers of different types. Other characteristics that may raise suspicion of a familial form of cancer susceptibility include a family history of cancer of the same type or related type in one or more firstdegree relatives, a high rate of cancer in the family, or cancer occurring in an individual or with a family with congenital anomalies or birth defects. Several hereditary cancer syndromes have been identified and are characterized by clustering of specific types of cancers within families. An online catalog of cancer family syndromes is available at www.ncbi.nlm.nih.gov in the Online Mendelian Inheritance in Man (OMIM) database.
Malignancies of the colon, breast, and ovary are the most common hereditary cancers in women, and approximately 5 to 10% of these cancers have a strong hereditary basis. In the 1990s, many of the genes responsible for high-penetrance hereditary cancer syndromes were identified, and this has facilitated genetic testing and clinical interventions aimed at decreasing cancer mortality.1, 2, 3 With the availability of genetic testing, cancer prevention and early detection efforts can be focused on mutation carriers. Noncarriers in these families can be reassured they are not at greater risk than the general population. This chapter summarizes progress to date in the diagnosis and management of hereditary cancer syndromes.
BASIC CANCER GENETICS
Cancer is a disease in which there is loss of growth regulatory control, allowing unrestrained proliferation of tumor cells. Normal cellular proliferation is regulated by complex molecular pathways that contain numerous checks and balances. Oncogenes encode proteins that stimulate proliferation, whereas tumor suppressor genes encode proteins that inhibit proliferation. In each organ, there is a carefully programmed balance between these opposing classes of gene products. In some organs, such as the bone marrow, the genetic program allows for constant proliferation and regeneration of the cellular supply; whereas in the brain, proliferation rarely occurs and the total number of cells is relatively static. Some of these oncogenes and tumor suppressor genes are also involved in regulating differentiation and apoptosis (programmed cell death), which are processes that also serve to control the population of cells within an organ.
Alterations in the genes that control cellular growth can cause malignant transformation. Oncogenes can be activated by several mechanisms. In some types of cancers, activation occurs by amplification of oncogenes (e.g., c-myc, HER-2/neu). Instead of 2 copies of one of these growth stimulatory genes, there may be as many as 40 copies. Some oncogenes may become overactive if affected by point mutations (e.g., K-ras). Finally, oncogenes may be translocated from one chromosomal location to another, and they may come under the influence of promoter sequences that cause overexpression of the gene (e.g., abl).
Loss of tumor suppressor gene function can also result in overactive proliferation and outgrowth of a tumor. This usually involves a two-step process in which both copies of a given tumor suppressor gene are inactivated. In most cases, mutation of one copy of a tumor suppressor gene occurs, along with complete loss of the other copy of the gene due to deletion of a large segment of the chromosome where the gene resides. Knudson4 termed this process the “two-hit” hypothesis of cancer development. DNA repair genes play an important role in preventing the development of human cancers. Each time a cell divides, the DNA is copied so both cells will receive a complete set of the genetic code. Although DNA synthesis occurs with high fidelity, it is estimated that spontaneous errors (mutations) occur about once every million bases. In nondividing cells, mutations may arise due to endogenous processes such as DNA methylation and deamination. The cellular DNA repair systems are able to fix much of this genetic damage, but some mutations elude this surveillance system. The rate at which critical mutations occur in cells may be accelerated if the DNA repair genes themselves have undergone inactivating mutations. In general, cancer occurs only after damage to several DNA repair genes and growth regulatory oncogenes and tumor suppressor genes.
Hereditary cancers arise due to an inherited mutation in one of the earlier mentioned classes of genes (see Table 21.1 for some selected hereditary cancer syndromes). Persons who inherit a mutation in a cancer gene carry that mutation in every cell of their body and have a strikingly increased lifetime risk of developing one or more cancers when the second copy of the gene is inactivated in a single cell. These cancers often develop at a younger age than would be expected, including in children. Some rare cancers in young children, such as retinoblastoma and Wilms tumor, result from inherited mutations in tumor suppressor genes.
PRINCIPLES OF CANCER GENETIC COUNSELING
Obtaining a thorough cancer history from the patient and/or the family is a vital first step in caring for families with hereditary cancer. The seemingly simple act of gathering information forms the foundation on which the rest of the process is based. The importance of careful documentation of family history, including review of clinical materials from other cancer cases in the family, has been stressed for many years by Dr. Henry T. Lynch, one of the pioneers of hereditary cancer genetics.5 His own compulsively assembled pedigrees allowed him to define two familial cancer syndromes: Lynch I (colon cancer), and Lynch II (colon cancer and other cancers).5
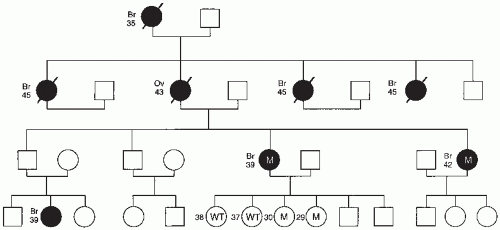
Family history questionnaires administered in an office setting are an effective method of eliciting an initial family history.6 A complete cancer family history should begin by ascertaining cancer information, including age of onset, and specific type of cancer in all first-degree relatives, including mother, father, siblings, and children. Data on second-degree relatives, including grandparents, aunts, and uncles, should also be obtained from both maternal and paternal sides. Family members without cancer should be included because their unaffected status can provide clues to the inheritance pattern. A family cancer history should ideally be obtained from more than one individual in families where there is a suspicion of a hereditary syndrome.7 This information can be depicted diagrammatically in a pedigree (Fig. 21.1). To make a pedigree as accurate as possible, cancer diagnoses should be confirmed when possible by review of pathologic specimens or medical records.
Pedigrees concisely convey a large amount of information, including the presenting cancer case (proband), others with cancer, cancer decedents, and potential mutation carriers. Important additional information recorded includes the age of onset of cancers, second cancers, bilaterally of a cancer, stage of the cancer, and metastatic location. In addition, cancer histology, age of unaffected individuals, exposure to carcinogenic agents, suspected extended family history of cancers, and the pattern of cancer occurrence among generations is recorded.8 A known genetic disorder in the family should be prominently noted because some syndromes also predispose individuals to cancers (e.g., Fanconi anemia, xeroderma pigmentosa, Bloom syndrome, ataxia-telangiectasia). In addition to family history, a complete personal history of potential cancer risk-modifying factors should be obtained, for example, hormone use, birth control pill use, menopausal status, diet, parity, breast-feeding history, and concomitant medical conditions.
FIGURE 21.1 Familial ovarian cancer pedigree with BRCA1 mutation. The ages of family members and types of cancer are noted. Slashes denote individuals who have died of cancer. Individuals denoted M are BRCA1 mutation carriers, whereas individuals denoted WT have normal BRCA1. □, males; ○, females; ▪, affected males; ●, affected females.
An ethnic history should also be elucidated, especially for patients of Ashkenazi (Eastern and Central European, including Germany, Poland, Lithuania, Ukraine, and Russia) Jewish heritage. Specific mutations, called founder mutations, that are fairly common have been identified in various ethnic groups.9 It is thought that founder mutations arose many generations ago in a single individual and later became more prevalent, perhaps because the ancestral groups was once geographically or culturally isolated, or dramatically contracted. The most common founder mutations described thus far are the BRCA1 185delAG and BRCA2 6174delT mutations, which occur in about 1.0 and 1.4% of Ashkenazi Jews, respectively.10,11 A third less common founder mutation (BRCA1 5382insC) also has been noted in Ashkenazi populations. These three mutations account for 98 to 99% of the BRCA1/BRCA2 mutations identified in Ashkenazi Jews.9 Today, most of the Ashkenazi Jews live in the United States, Israel, South America, South Africa, Australia, and New Zealand. One Israeli study, with 199 Ashkenazi women and 44 non-Ashkenazi women, found that about half of women with ovarian cancer and one-third of women with breast cancer had one of the three Ashkenazi founder mutations.12 Certain founder mutations have also been identified in populations from the Netherlands, Sweden, Hungary, Iceland, and French Canada.
Male breast cancer cases are likely to harbor an inherited BRCA2 mutation and should be specifically included in the assembled pedigree.12, 13, 14 The ratio of female-to-male births in an affected family may also provide important information about a hereditary cancer syndrome.12,13,15BRCA1 and BRCA2 mutations appear to statistically decrease the number of male offspring in mutation carriers, while not affecting overall fertility.16
Once a family is suspected of having a hereditary cancer syndrome, genetic counselors should be involved in all phases of the genetic risk assessment and testing process.3 A list of trained counselors and their contact information in the Unites States is available on the National Cancer institute (NCI) website at http://www.cancer.gov/cancertopics/genetics/directory. Extensive nondirective counseling and education is essential prior to genetic testing. Individuals should understand the risk of testing positive, potential benefits of the test, and the social and psychological implications of testing (see Table 21.2 NOW 2 for a summary of the benefits and limitations of genetic testing). The American Society of Clinical Oncology has outlined the basic elements of informed consent for genetic testing for cancer susceptibility, which should be met prior to ordering a test.17 For these reasons, direct-to-consumer genetic testing has been discouraged by leading professional societies and bioethicists.17, 18, 19, 20 If direct-to-consumer testing is performed, the companies involved have an obligation to ensure that appropriate counseling occurs.
Several algorithms have been developed for risk assessment of the likelihood of carrying a BRCA1 or BRCA2 mutation, including BOADICEA, BRCAPRO, IBIS, Myriad and the Manchester scoring system.21 Of these, the BOADICEA algorithm appears to be the most accurate.21 After pedigree analysis and risk assessment, women who have a 20 to 25% risk of harboring a genetic mutation should be offered genetic testing.3 This group includes women with a personal history of both breast and ovarian cancer, women with ovarian cancer and a family history of premenopausal breast cancer, women in Ashkenazi families with early onset breast cancers, women with male breast cancer in relatives, and women in known BRCA families.3
TABLE 21.2 Benefits and Limitations of Genetic Testing
Possible Benefits and Limitations Associated With Genetic Testinga
The potential benefits of testing include the following:
Reassurance and reduced uncertainty.
Facilitation of informed decision making about cancer prevention, including risk-reducing surgery and therapy for an existing breast cancer.
Testing may satisfy a desire to obtain information for other family members, particularly one’s children.
Test results may help guide reproductive plans or choices.
Limitations and/or risks of testing include the following:
A positive test result for a known susceptibility mutation indicates that the individual has an increased risk of developing cancer; however, it is not inevitable that a cancer will develop.
Uninformative result or a “variant of uncertain significance.”
The psychosocial impact may be considerable. A common reason for declining genetic testing is the fear of breach of privacy and genetic discrimination in the areas of health and life insurance or employment.
a aA positive results means that a deleterious mutation was identified. A true negative result occurs when an individual tests negative for a mutation that has already been identified in his or her family. An uninformative result occurs when no deleterious mutations are found in a high-risk individual who is the first to be tested in a family. This may occur when a deleterious mutation or rearrangement is present but cannot be detected by available methods; another, a rare gene or one that has not been isolated yet is involved; or the individual being tested developed sporadic rather than hereditary cancer. A “variant of uncertain significance” is an uninformative result as the significance of the variant is not well established; in these cases, further testing of family members may be helpful.
A second group of women with an inherited risk of 5 to 10% can be considered for genetic testing. This group includes women with premenopausal breast cancer; women who have personal histories of gynecologic malignancies, including ovarian, fallopian tube, or peritoneum; bilateral breast cancer survivors; Ashkenazi Jewish women with familial breast cancers in the 40s and 50s; women breast cancer survivors younger than age 50 years with other family history of breast cancer at age younger than 50 years, or multiple cases of breast cancer.3 A list of testing laboratories is available through the Gene Tests website at www.genetests.org.
The use of predictive models to ascertain the likelihood of finding a BRCA mutation can be cumbersome for clinicians. As a result, consensus guidelines with criteria for genetic testing have been developed by several groups, including the American College of Obstetricians Gynecologists (ACOG) and the National Comprehensive Cancer Network (NCCN) (see Tables 21.3 and 21.4).
Testing should be offered to both women and men in a high-risk family.22,23 In the United States, most insurance companies will cover the majority of the costs of commercial BRCA testing in appropriate candidates, although a letter of medical necessity stating the potential impact of a positive test result on surveillance or possible surgical options may be needed. Most individuals who seek genetic counseling elect to undergo cancer genetic testing. If possible, a family member who has already had cancer should be tested first. If a mutation is found in an affected individual, this greatly facilitates testing of other family members for this specific mutation. Test results should be conveyed in a setting that includes multidisciplinary input from genetic counselors and clinicians. Additional psychological support and encouragement is offered at this juncture because both positive and negative test results can cause significant distress.24 The lifetime cancer risk for mutation carriers, as well as the options for prevention, should be reviewed. Patient disclosure of their genetic testing results to other potentially affected family members should be discussed and encouraged, but the decision to disclose those results remains with the patient.22 Some families with a strong cancer history do not carry a mutation in a known susceptibility gene. Referral of these families to institutions with expertise in cancer genetics will assist in the identification of other cancercausing genes through more intensive genetic analysis and ongoing research projects. In the meantime, clinical decisions in such families are made on a case-by-case basis. In some cases, preventive strategies, including surgical prophylaxis, may be warranted even when genetic testing does not reveal an alteration in a known cancer susceptibility gene.
TABLE 21.3 Criteria for Genetic Risk Assessment
Patients with greater than an approximate 20-25% chance of having an inherited predisposition to breast cancer and ovarian cancer and for whom genetic risk assessment is recommended:
Women with a personal history of both breast cancer and ovarian cancera
Women with ovarian cancera and a close relativeb with ovarian cancer or premenopausal breast cancer or both
Women with ovarian cancera who are of Ashkenazi Jewish ancestry
Women with breast cancer at age 50 years or younger and a close relativeb with ovarian cancera or male breast cancer at any age
Women of Ashkenazi Jewish ancestry in whom breast cancer was diagnosed at age 40 years or younger
Women with a close relativeb with a known BRCA1 or BRCA2 mutation
Patients with greater than an approximate 5-10% chance of having an inherited predisposition to breast cancer and ovarian cancer and for whom genetic risk assessment may be helpful:
Women with breast cancer at age 40 years or younger
Women with ovarian cancer, primary peritoneal cancer, or fallopian tube cancer of high grade, serous histology at any age
Women with bilateral breast cancer (particularly if the first case of breast cancer was diagnosed at age 50 years or younger)
Women with breast cancer at age 50 years or younger and a close relativeb with breast cancer at age 50 years or younger
Women of Ashkenazi Jewish ancestry with breast cancer at age 50 years or younger
Women with breast cancer at any age and two or more close relativesb with breast cancer at any age (particularly if at least one case of breast cancer was diagnosed at age 50 years or younger)
Unaffected women with a close relativeb that meets one of the previous criteria
a aCancer of the peritoneum and fallopian tubes should be considered a part of the spectrum of the hereditary breast and ovarian cancer syndrome.
b bClose relative is defined as a first-degree relative (mother, sister, daughter) or second-degree relative (grandmother, granddaughter, aunt, niece).
From American College of Obstetrician and Gynecologists. ACOG Practice Bulletin No. 103: hereditary breast and ovarian cancer syndrome. Obstet Gynecol. 2009;113:957-966.
TABLE 21.4 National Comprehensive Cancer Network Criteria for Breast Cancer Genetics Referral
≥1 family member on same side of family with a combination of breast cancer and ≥1 of the following (especially if early onset): pancreatic cancer, aggressive prostate cancer (Gleason score ≥7); sarcoma, adrenocortical carcinoma, brain tumors, endometrial cancer, leukemia/lymphoma; thyroid cancer, dermatologic manifestations and/or macrocephaly, hamartomatous polyps of GI tractg; diffuse gastric cancerb
An unaffected individual with a family history of one or more of the followingf:
A known mutation in a breast cancer susceptibility gene within the family
≥2 breast primaries in single individual
≥2 individuals with breast primaries on the same side of family (maternal or paternal)
≥1 ovariane cancer primary from the same side of family (maternal or paternal)
First- or second-degree relative with breast cancer ≤45 y
≥1 family member on same side of family with a combination of breast cancer and ≥1 of the following (especially if early onset): pancreatic cancer, aggressive prostate cancer (Gleason score ≥7); sarcoma, adrenocortical carcinoma, brain tumors, endometrial cancer, leukemia/lymphoma; thyroid cancer, dermatologic manifestations and/or macrocephaly, hamartomatous polyps of GI tractg; diffuse gastric cancerb
Male breast cancer
a aThe criteria for further risk evaluation and genetic testing are not identical. For the purposes of these guidelines, invasive and ductal carcinoma in situ breast cancers should be included. The maternal and paternal sides of the family should be considered independently for familial patterns of cancer.
b bClinically use age ≤50 y because studies define early onset as either ≤40 or ≤50 y.
c cTwo breast primaries includes bilateral (contralateral) disease or two or more clearly separate ipsilateral primary tumors either synchronously or asynchronously.
d dClose blood relatives include first-, second-, and third-degree relatives.
e eFor the purposes of these guidelines, fallopian tube and primary peritoneal cancers are included. Ovarian/fallopian tube/primary peritoneal cancers are component tumors of Lynch syndrome/hereditary non-polyposis colorectal cancer; be attentive or clinical evidence of this syndrome.
f fFor populations at increased risk, requirements for inclusion may be modified (e.g., women of Ashkenazi Jewish descent with breast or ovarian or pancreatic cancer at any age).
g gFor hamartomatous colon polyps in conjunction with breast cancer and hyperpigmented macules of the lips and oral mucosa, STK11 testing should be considered.
h hFor lobular breast cancer with a family history of diffuse gastric cancer, CDH1 gene testing should be considered.
In studies of high-risk hereditary breast and ovarian cancer clinics, between 60 to 64% of women opt to be tested for genetic mutations.25,26 The most common reasons for declining testing include lack of health insurance, fear of insurance discrimination based on test results or simply having testing done, concerns about the accuracy of the tests, and the possible emotional impact of test results on the patient and her family.27
HEREDITARY OVARIAN AND BREAST CANCER GENETICS
Autosomal dominant hereditary syndromes account for about 5% of breast cancers and 10% of ovarian cancers. Women with hereditary breast and ovarian cancer syndrome have a lifetime risk of breast cancer of 50 to 85%.28,29 This wide range in reported risk is likely because of variations in risk due to penetrance and risk modifiers among the different study populations. Germline mutations in BRCA1 and BRCA2 account for the vast majority of high-penetrance hereditary ovarian cancers and at least half of hereditary breast cancers. These mutations may also be associated with increased risk for other malignancies (see Table 21.5). Populationbased studies have demonstrated that BRCA mutations are rare in the non-Ashkenazi general population, affecting approximately 1 in 800 to 1 in 1000 individuals.30,31 In the Ashkenazi Jewish population, the BRCA1/2 founder mutation carrier rate is estimated at 2.5%, affecting 1 in 40 Ashkenazi Jews.32,33 A variant in the CHEK2 gene (1100del C) has been identified that is present in about 1% of the population and this variant increases risk of breast cancer by about two-fold.34
Mutation carriers of the BRCA1 and BRCA2 genes have only one functional allele of these genes in their cells, not two. This results in an increase in genomic instability and tumorigenesis. It is not clear why these mutations predispose mainly to breast and ovarian cancer. There are over a thousand known mutations in BRCA1 and BRCA2 genes and most of these lead to a truncated protein when they are translated while others may result in an abnormal amount or conformation of the BRCA1 and BRCA2 protein(s).
Patients with BRCA1 or BRCA2 mutations who develop breast cancer have an increased risk of second breast cancers. The rates of contralateral breast recurrence in patients with BRCA-associated breast cancer at 5 and 10 years are 11 to 20% and 25 to 27%, respectively.35,36 The risk appears to be related to the patient’s age at the first diagnosis of breast cancer. Although not as well defined, there is an increased risk for ipsilateral breast cancer recurrence over time, with most of these representing second primary cancers rather than local recurrences.37
BRCA1
The BRCA1 gene is a very large gene of 7.8 kb, encoding a protein of 1863 amino acids, located on chromosome 17q21.38BRCA1 plays roles in repair of double-stranded DNA breaks, regulation of gene transcription and association with other cell signaling molecules not involved in DNA repair. Loss of BRCA1 function initiates the development of other genetic alterations through the inability to repair double-stranded DNA damage that eventually leads to clinically recognizable cancer.39
TABLE 21.5 Estimated Cancer Risks Associated With BRCA1 and BRCA2 Mutations
Cancer Type
Risk in BRCA1/2 Carriers to Age 70 Years
General Population Risk to Age 70 Years
Comments
Breast
40 to 75%
7%
The range of risk reported in the literature is wide. In most studies, risk in BRCA1 carriers is higher than that observed in BRCA2 carriers.
The incidence of breast cancer diagnosed younger than 50 years of age is higher in BRCA1 carriers compared to BRCA2 carriers, but both groups have an increased risk of premenopausal breast cancer, as well as increased lifetime risks.
Contralateral (opposite) breast
BRCA1: up to 65%
BRCA2: up to 50%
0.5 to 1% per year after diagnosis
Risk is affected by other factors such as tamoxifen use and oophorectomy (ovary removal).
For BRCA1/2 carriers who have had lumpectomy: Risk of developing a second breast cancer in the affected breast appears to be elevated over long follow-up periods.
Ovarian
BRCA1: Approximately 40%
BRCA2: Approximately 15%
<1%
The risk estimates provided here are representative of findings from multiple studies.
The incidence of ovarian cancer diagnosed younger than 50 years of age is higher in BRCA1 carriers, and overall rare in all carriers younger than 40 years old; risk of fallopian tube cancer is also substantially elevated.
Colon
Unclear
2%
If elevated, risk is small; studies have not been consistent about whether risk is elevated.
Prostate
Elevated; absolute risk not well defined
8% Whites 12% African Americans
Risk appears to be higher in BRCA2 carriers and possibly in men younger than 65 years old.
Male breast
Elevated but <10%
<1%
Risk appears to be higher in BRCA2 carriers; rarely occurs in men younger than age 50.
Pancreatic
Elevated but <10%
<1%
Risk appears to be higher in BRCA2 carriers.
Other bites
To be determined
Varied
These sites may include cancer of the stomach and skin (melanoma).
These risks are estimates based on review of the literature. Specific studies have reported risks that are lower or higher than the ranges or estimates quoted; however, the estimates reported here are representative of findings from high-quality studies. Risks will also vary based on an individual’s current age and other risk factors.
Reproduced with permission from Carlson KJ. Screening for ovarian cancer. UpToDate Web site. http://www.uptodate.com/contents/screening-for-ovarian-cancer. Accessed November 26, 2013.
The lifetime risk of breast cancer by age 70 years in BRCA1 mutation carriers is about 65 to 75%.40 The lifetime risk (by age 70 years) of ovarian cancer in BRCA1 carriers is now believed to be as high as 39 to 46%, which is higher than for BRCA2 mutation carriers.40 In addition, some studies have suggested that BRCA1 mutation carriers have a higher risk of prostate, colon, pancreatic, and stomach cancers.13,41 A more recent study of Ashkenazi Jewish carriers confirmed the increased risk of prostate cancer, but colon cancer risk was not increased.13,32 Pancreatic cancer risk is elevated in BRCA1 as well as BRCA2 carriers.42 Increased breast cancer risk begins earlier than risk for ovarian cancer.41 Carriers have an increased breast cancer risk in their twenties, whereas their ovarian cancer risk does not rise appreciably until the mid-thirties. In addition to risk of an initial breast cancer, BRCA1 mutation carriers are at about a 40% risk of developing a second primary breast cancer by the age of 70 years.13
Various BRCA1 mutations may differ in the extent to which they predispose to breast or ovarian cancer. As a result, individuals in the same family may carry the same mutation but have significantly different types of cancer and even ages of onset. Mutations in the carboxy terminus of the gene may result in a higher frequency of breast cancer relative to ovarian cancer.43, 44, 45 Conversely, ovarian cancer is associated in some studies with mutations in the middle of the gene, the area called the ovarian cancer cluster region, between bases 2401 and 4190.46 However, these findings are not consistent enough to guide clinical management or recommendations.
Differences in penetrance have also been noted among families with identical BRCA1 mutations. Two hypotheses, both of which may be relevant, have been proposed to explain variable penetrance. First, other gene variants, including single nucleotide polymorphisms (SNPs), may modify the penetrance of BRCA1, that is, gene-gene interactions may contribute to differences in cancer expression. Alternatively, it is possible that gene-environment interactions may modify risk. For example, pregnancy and oral contraceptive pill use decrease the risk of ovarian cancer in the general population. Reproductive risk factors that affect breast and ovarian cancer incidence in the general population may also modify risk in BRCA1 carriers. In some studies, oral contraceptives were found to reduce ovarian cancer risk by 50% in women with hereditary ovarian cancer syndromes.47 These two hypotheses are not mutually exclusive, because BRCA1 itself has been found to modify estrogen receptor α.48
BRCA1-associated breast cancers cluster in the basallike gene expression pattern.49BRCA1-associated breast tumors are typically estrogen and progesterone receptor negative, as are basal cancers. Amplification of HER2/neu is uncommon in BRCA1-associated tumors. BRCA1-associated breast cancers have other high-risk features, including high-grade, high proliferative rate, no special type (NST) ductal carcinoma histology, and medullary histology.50 They also have lymphocyte and plasma cell infiltrates. Despite these poor prognostic features, BRCA1-associated breast cancers presented at lower stage and have a relatively favorable outcome.51 The addition of chemotherapy to conservative surgery for breast cancer increased the survival of Ashkenazi women with BRCA mutations in one study.52
The histologic features of ovarian cancers in BRCA1 carriers do not differ strikingly from sporadic cancers. Most cases are advanced stage, moderate to poorly differentiated serous cancers. BRCA1-associated high-grade serous ovarian cancers are histologically indistinguishable from fallopian tube or primary peritoneal cancers, which also are more common in BRCA1 carriers.13,53, 54, 55, 56 The finding that the spectrum of BRCA1-associated serous cancers includes the tubal epithelium has led some to hypothesize that the fallopian tube is the primary site of origin of ovarian and primary peritoneal serous carcinomas.57,58 This theory is further supported by the finding of intraepithelial carcinomas in the tubal fimbria of some prophylactic surgery specimens from mutation carriers.59, 60, 61, 62 In some cases, tubal in situ carcinomas (TICs) have been found adjacent to invasive disease, which provides further evidence that they are precursor lesions. TICs have been shown to overexpress mutant p53, as do most high-grade serous cancers.63,64
Survival of BRCA1 carriers with ovarian cancer is longer than sporadic cases matched for age, stage, and other prognostic factors.65, 66, 67, 68, 69 This is thought to be due to a better response to platinum-based chemotherapy because of loss of BRCA1-associated repair of doublestranded DNA repair.69,70 It also has recently been shown that agents that inhibit the enzyme polyADP-ribose polymerase (PARP), which is involved in single-stranded DNA repair, are highly active in BRCA1-associated cancers, but not in sporadic cases.71, 72, 73, 74 Abrogation of both single- and double-stranded DNA repair in these cancers makes them acutely sensitive to DNA damaging chemotherapy agents. Acquisition of platinum resistance in BRCA mutant tumors may be associated with secondary genetic changes in BRCA1 that restored full-length protein expression and function.75
BRCA2
A significant fraction of breast cancer families in which a BRCA1 mutation was excluded were found to be linked to chromosome 13q12 in 1994.76BRCA2 binds and regulates the strand invasion recombinase, which is necessary for proper recombination of double-strand DNA breaks. In 1995, this second breast cancer susceptibility gene (BRCA2) was identified.77 Notable attributes of BRCA2 families include early age of onset, most often in the last 40s or 50s, and the occurrence of male breast cancer.78,79 Ovarian cancer is a less prominent feature of these families, and there are about 10 female breast cancer cases and 1 male breast cancer case for each ovarian cancer case in BRCA2 families. Fallopian tube cancer risk is also increased.53,54,56
BRCA2 is responsible for a similar proportion of hereditary breast cancer as BRCA1 (4 to 5%), and studies indicate that mutations in BRCA2 confer a similar lifetime risk of female breast cancer as for BRCA1. The current estimated lifetime risk of ovarian cancer in BRCA2 carriers is between 10 and 20%.32,54,80,81 Ovarian cancer is most often observed in families with mutations in the large exon 11 in the Ovarian Cancer Cluster Region (OCCR).9,82 Similar to BRCA1, the BRCA2 gene appears to play a role in double-stranded DNA repair.83BRCA2 is now known to be identical with a gene that causes some cases of Fanconi anemia.84
The clinicopathologic characteristics of BRCA2-associated breast cancers are similar to sporadic cases, but BRCA2-associated cases are less often tubular and are of higher grade.85BRCA2-associated cancers generally are of the luminal subtype and are often estrogen and progesterone receptor positive.49BRCA2-associated ovarian cancers are usually high-grade serous lesions, although some may be endometrioid. The risk of clear cell and mucinous cancers and borderline tumors is not increased in BRCA1/2 carriers. Carriers of the BRCA2 mutations are also at increased risk of other cancers, including prostate, laryngeal, and pancreatic cancer. Despite multiple cancer risks, women with BRCA2 mutations have an increased life expectancy compared to BRCA1 carriers, due to the increased rate and mortality from ovarian cancer in BRCA1 carriers.86
Genetic Testing for Hereditary Ovarian and Breast Cancer
Mutations have been observed throughout the large BRCA1 and BRCA2 genes, and approximately 80 to 90% of the mutations predict truncated protein products.87, 88, 89, 90 Missense mutations alter a single amino acid in a fulllength protein product and occur in about 10 to 15% of hereditary cases.88,89 Alteration of a single amino acid may create a disease-causing mutation or may not affect gene function.38 In some cases, biochemical and computational analyses73,90, 91, 92, 93 of variants of uncertain significance have helped to clarify whether they represent clinically significant deleterious changes.94, 95, 96, 97, 98, 99 Segregation of a missense mutation with breast and ovarian cancer in a family suggests, but does not prove, its clinical significance.
Because mutations in BRCA1 and BRCA2 occur throughout these large genes, the most reliable method of detecting mutations is to sequence the entire coding region, including intronic splice sites. The sensitivity of DNA sequencing for detecting mutations is excellent and the false-negative rate is estimated to be very low. Although automated DNA sequencing is labor intensive and expensive, it remains the gold standard for mutational testing. However, some deleterious BRCA1/2 mutations involve complex rearrangements of the gene that do not result in a sequence change, yet a functional protein product is not produced. Testing for these rare rearrangements with BRCA1/2 Analysis Rearrangement Test (BART) analysis is appropriate in families with a very high predicted likelihood of carrying a BRCA1/2 mutation in which no mutation is found in the coding sequence.100,101
Although mutations in BRCA1 and BRCA2 have been noted in some women in the absence of a family history of breast or ovarian cancer, the incidence is low and cost considerations prohibit BRCA mutational screening in the general population of the United States. BRCA1/2 testing costs about $3,000. A postmenopausal woman with breast or ovarian cancer who does not have a family history of ovarian or breast cancer has a 3% risk of carrying a mutation. At the other extreme, in families with two cases of breast cancer and two cases of ovarian cancer, the probability of finding a mutation may be as high as 80 to 90%.45 Most experts believe it is reasonable to offer testing when the family history suggests at least a 5 to 10% probability of finding a mutation.3 In practical terms, this translates into two first-degree relatives with either ovarian cancer at any age, breast cancer prior to age 50 years, and those in a high-risk ethnic group such as Ashkenazi Jews. In some families with a paucity of females, it may be reasonable to recommend testing in families with fewer cancer cases.102
Within the Ashkenazi Jewish population, 1 in 40 individuals carry one of three germline BRCA1 or BRCA2 mutations.30,80,81,103,104 Testing for these three founder mutations is much less expensive than full sequencing, and some believe this should be offered to all Ashkenazi women with breast and/or ovarian cancers.102,105,106 It also has been suggested that perhaps all women with high-grade serous ovarian cancers, particularly those who have a family history of breast and ovarian cancer, should undergo full BRCA1/2 testing. The mutation rate may be as high as 20% in this subgroup, and those found to be mutation carriers can be predicted to have a better prognosis and to respond well to platinum drugs and PARP inhibitors.5,72,74 As the cost of DNA sequencing continues to fall in the future, this may facilitate more widespread genetic testing in women with ovarian cancer.
Only gold members can continue reading. Log In or Register to continue