Chapter 600 Developmental Disorders of Muscle
Myogenic Regulatory Genes and Genetic Loci of Inherited Diseases of Muscle
A family of four myogenic regulatory genes shares encoding transcription factors of “basic helix-loop-helix” (bHLH) proteins associated with common DNA nucleotide sequences (Table 600-1). These genes direct the differentiation of striated muscle from any undifferentiated mesodermal cell. The earliest bHLH gene to program the differentiation of myoblasts is myogenic factor 5 (Myf5). The second gene, myogenin, promotes fusion of myoblasts to form myotubes. Herculin (also known as MYF6) and MYOD1 are the other two myogenic genes. Myf5 cannot support myogenic differentiation without myogenin, MyoD, and MYF6. Each of these four genes can activate the expression of at least one other and, under certain circumstances, can autoactivate as well. The expression of MYF5 and of herculin is transient in early ontogenesis but returns later in fetal life and persists into adult life. The human locus of the MYOD1 gene is on chromosome 11, very near to the domain associated with embryonal rhabdomyosarcoma. The genes encoding Myf5 and herculin are on chromosome 12 and that for myogenin is on chromosome 1.
Table 600-1 INHERITANCE PATTERNS AND CHROMOSOMAL OR MITOCHONDRIAL LOCI OF NEUROMUSCULAR DISEASES AFFECTING THE PEDIATRIC AGE GROUP
| DISEASE | TRANSMISSION | LOCUS |
|---|---|---|
| Duchenne and Becker muscular dystrophy | XR | Xp21.2 |
| Emery-Dreifuss muscular dystrophy | XR | Xq28 |
| Myotonic muscular dystrophy (Steinert) | AD | 19q13 |
| Facioscapulohumeral muscular dystrophy | AD | 4q35 |
| Limb-girdle muscular dystrophy | AD | 5q |
| Limb-girdle muscular dystrophy | AR | 15q |
| Congenital muscular dystrophy with merosin deficiency | AR | 6q2 |
| Congenital muscular dystrophy (Fukuyama) | AR | 8q31-33 |
| Myotubular myopathy | XR | Xq28 |
| Myotubular myopathy | AR | Unknown |
| Nemaline rod myopathy (NEM1) | AD | 1q21-q23 |
| Nemaline rod myopathy (NEM2) | AR | 2q21.2-q22 |
| Nemaline rod myopathy (NEM3) | AD, AR | 1q42.1 |
| Nemaline rod myopathy (NEM4) | AD | 9q13 |
| Nemaline rod myopathy (NEM5) | AR | 19q13 |
| Congenital muscle fiber-type disproportion | AR, X-linked R | 19p13.2, Xp23.12-p11.4, Xq13.1-q22.1; t(10; 17); sporadic |
| Central core disease | AD | 19q13.1 |
| Myotonia congenita (Thomsen) | AD | 7q35 |
| Myotonia congenita (Becker) | AR | 7q35 |
| Paramyotonia congenita | AD | 17q13.1-13.3 |
| Hyperkalemic periodic paralysis | AD | 17q13.1-13.3 |
| Hyperkalemic periodic paralysis | AD | 1q31-q32 |
| Glycogenosis II (Pompe; acid maltase deficiency) | AR | 17q23 |
| Glycogenosis V (McArdle; myophosphorylase deficiency) | AR | 11q13 |
| Glycogenosis VII (Tarui; phosphofructokinase deficiency) | AR | 1cenq32 |
| Glycogenosis IX (phosphoglycerate kinase deficiency) | XR | Xq13 |
| Glycogenosis X (phosphoglycerate mutase deficiency) | AR | 7p12-p13 |
| Glycogenosis XI (lactate dehydrogenase deficiency) | AR | 11p15.4 |
| Muscle carnitine deficiency | AR | Unknown |
| Muscle carnitine palmityltransferase deficiency 2 | AR | 1p32 |
| Spinal muscular atrophy (Werdnig-Hoffmann; Kugelberg-Welander) | AR | 5q11-q13 |
| Familial dysautonomia (Riley-Day) | AR | 9q31-33 |
| Hereditary motor-sensory neuropathy (Charcot-Marie-Tooth; Dejerine-Sottas) | AD | 17p11.2 |
| Hereditary motor-sensory neuropathy (axonal type) | AD | 1p35-p36 |
| Hereditary motor-sensory neuropathy (Charcot-Marie-Tooth-X) | XR | Xq13.1 |
| Mitochondrial myopathy (Kearns-Sayre) | Maternal; sporadic | Single large mtDNA deletion |
| Mitochondrial myopathy (MERRF) | Maternal | tRNA point mutation at position 8344 |
| Mitochondrial myopathy (MELAS) | Maternal | tRNA point mutation at positions 3243 and 3271 |
AD, autosomal dominant; AR, autosomal recessive; MELAS, mitochondrial encephalopathy lactic acidosis, and stroke; MERRF, myoclonic epilepsy with ragged-red fibers; mtDNA, mitochondrial deoxyribonucleic acid; tRNA, transfer ribonucleic acid; XR, X-linked recessive.
600.1 Myotubular Myopathy
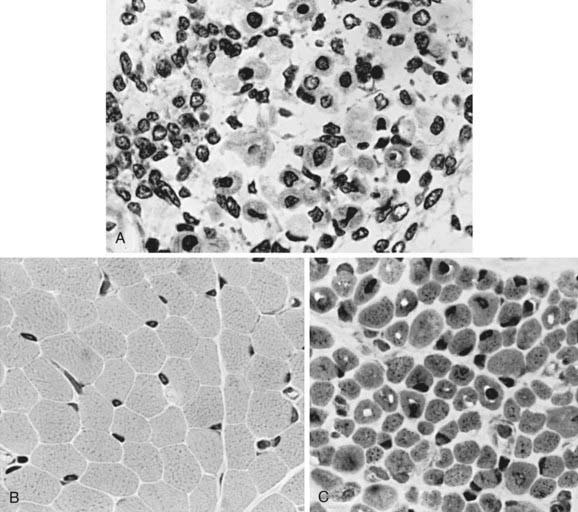
The term myotubular myopathy implies a maturational arrest of fetal muscle during the myotubular stage of development at 8-15 wk of gestation. It is based on the morphologic appearance of myofibers: A row of central nuclei lies within a core of cytoplasm; contractile myofibrils form a cylinder around this core (Fig. 600-1). Many challenge this interpretation and use the more neutral term centronuclear myopathy when referring to this myopathy. This term is nonspecific because internal nuclei occur in many unrelated myopathies.
Pathogenesis
The defective gene of the X-linked form and 3 genes of the autosomal recessive form are known.



