Developmental and Positional Anomalies of The Kidneys
Renal Embryology
The pronephros, which has no adult function, induces the mesonephros to differentiate into the mesonephric duct during the fourth to eighth week of fetal life. The mesonephric duct is the basis of the Wolffian system, which develops into the seminal vesicles, vas deferens, epididymis, and efferent ductules of the testis in boys, and the epoophoron and paraophoron (vestigial remnants between the fallopian tube and ovary) in girls. Between weeks 9 and 12, the ureteric bud branches off the mesonephric duct, contacts the metanephric blastema bud, and induces the entire collecting system of ureter, renal pelvis, calyx, and collecting tubules. The kidney develops via induction of the metanephric blastema by the ureteric bud into Bowman’s capsule, the convoluted tubules, and the loop of Henle.1 Figure 53-1 illustrates the progression of development from pronephros, to mesonephros, to metanephros.
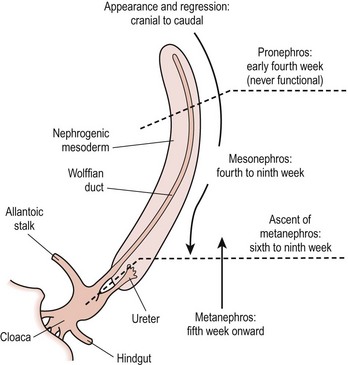
FIGURE 53-1 Development of the kidney. (Redrawn from Gray SW, Skandalakis JE. Embryology for Surgeons. Philadelphia: WB Saunders; 1972. p. 444.)
The kidneys begin at the upper sacral level with the renal pelvis facing anteriorly. The kidneys ascend either because the lumbar and sacral regions grow faster than the cervical and thoracic regions between 4 to 8 weeks, or because there is active migration. As the kidneys ascend, the renal pelvis rotates medially by 90°, leading to the normal configuration of the renal pelvis lying medial to the parenchyma. During this time, the blood supply shifts from inferior branches of the aorta to more cephalad branches, with the final renal artery being located at about L2. Failure of normal ascent leads to the persistence of a low-lying blood supply.1
Renal Dysplasia and Hypoplasia
Since the development of the kidney depends on proper interaction between the ureteric bud and the metanephric blastema, it should not be surprising that an abnormality in the location of the ureteral orifice is associated with abnormally induced renal tissue.2 Examination of the thickness of the renal parenchyma and number of glomeruli associated with normal and ectopic ureters in fetal specimens suggests that it is the initial interaction between bud and blastema, rather than subsequent obstruction or VUR, that determines if normal renal tissue will develop.2 Figure 53-2 shows how a ureter which arises in the proper trigonal location (A, E, F) is associated with normal renal parenchyma whereas a ureter arising from a more cranial location (B, C, D) or caudal location (G, H) is associated with progressively less normal renal parenchyma.
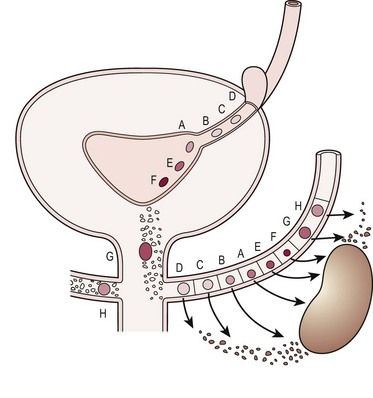
FIGURE 53-2 Relation of ureteral orifice location and associated metanephric tissue. (Redrawn from Mackie GG, Stephens FD. Duplex kidneys: A correlation of renal dysplasia with position of the ureteral orifice. J Urol 1975;114:274–80.)
Renal dysplasia and hypoplasia can be considered errors in renal induction. Figure 53-3 shows varying changes from agenesis to dysplasia and hypoplasia of the kidney. Although dysplasia is technically a histologic term, it refers to kidneys which contain primitive tubules either focally or diffusely. These ducts are lined by epithelium and surrounded by swirls of primitive collagen. No treatment is necessary for the dysplastic kidney, but there is an increased risk of reflux in the contralateral kidney.3 Hypoplastic kidneys are small, normal kidneys with a decreased number of nephrons. Dysplasia can also occur in hypoplastic kidneys. While secondary hypoplasia can occur due to infection or obstruction, two types of hypoplastic kidneys are clinically important: the oligomeganephronic type, and the Ask–Upmark kidney. In oligomeganephronia, there is a decrease in the number of nephrons with an associated hypertrophy of the ones which are present. Patients present with polyuria and failure to concentrate their urine, but no hypertension. Imaging with ultrasound (US) reveals small kidneys. Medical management with protein restriction, and high fluid and salt intake is initiated. Once the glomerular filtration rate drops significantly, dialysis is required.4 The Ask–Upmark kidney was initially felt to be a developmental problem, but is now believed to represent reflux nephropathy. The key finding is a small kidney with segmental hypoplasia, probably secondary to ascending pyelonephritis. VUR and hypertension are usually present. Most patients are over 10 years of age with a 2 : 1 female : male ratio. If the disease is unilateral, nephrectomy may cure the hypertension. Bilateral disease is managed medically.5
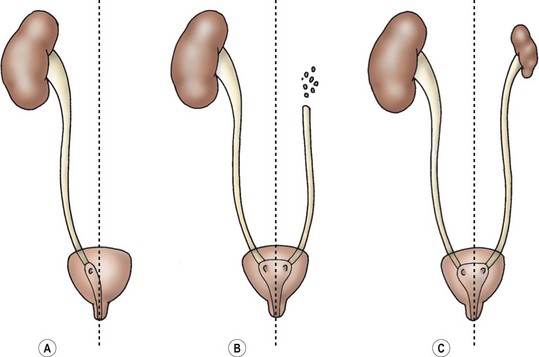
Renal Agenesis
Unilateral renal agenesis occurs in 1 : 1,000 live births with a 2 : 1 male predominance.6,7 Unilateral renal agenesis can result in compensatory hypertrophy of the contralateral kidney. The left kidney is more likely to be affected in unilateral renal agenesis.8 Since unilateral renal agenesis is asymptomatic and eventual renal function is normal, the diagnosis is usually made on prenatal ultrasound, or it is incidentally found during imaging for other abdominal symptoms. Sometimes it can be suspected on plain abdominal films if the colon is medially deviated at the splenic or hepatic flexures.9 These patients should consider obtaining a medical alert bracelet so that in case of traumatic injury, the solitary kidney is not inadvertently removed.
In a newborn with the prenatal diagnosis of unilateral renal agenesis, physical examination at the time of birth should be focused on detecting the anomalies present in the VACTERL association (Box 53-1).10 A voiding cystourethrogram (VCUG) should also be obtained since approximately 30% of VACTERL patients with unilateral renal agenesis will have VUR in the contralateral kidney.10
Females with unilateral renal agenesis should have their genital anatomy evaluated since up to 30% will have an abnormality of the Müllerian duct due to the Mayer–Rokitansky syndrome (Müllerian, uterine, upper vaginal duplications with or without obstruction, or vaginal agenesis).11,12 The abnormal induction of the mesonephric duct is believed to cause partial or complete nonunion of the paired Müllerian ducts.13 Conversely, 40% of patients with abnormalities of the Müllerian organs will have unilateral renal agenesis or ectopia.14 In patients with duplicated vaginas and unilateral vaginal agenesis, the side without a vagina is also the side without a kidney.13
If the diagnosis of Mayer–Rokitansky is not made prenatally, the patients can present either as infants with hydrocolpos, or as adolescents with lower abdominal pain after the onset of menses due to an obstructed vagina or uterus (with or without duplication). Magnetic resonance imaging (MRI) is useful in delineating the pelvic anatomy in these cases. In vaginal agenesis, the vagina is only present as a shallow pouch. There is a wide variety of abnormalities of the vagina, uterus, and fallopian tubes (Fig. 53-4), but the ovaries are embryologically normal.
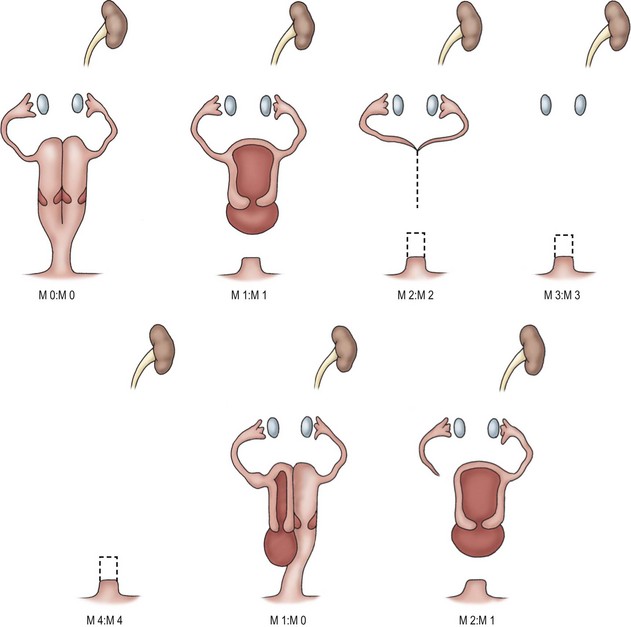
FIGURE 53-4 Variations in Müllerian anatomy in Mayer–Rokitansky syndrome. M0, Right or left vagina and uterus, or duplex vagina and uterus with partial or complex septum. M1, Partial or complete absence of vagina. M2, Absence of vagina and uterus. M3, Absence of vagina, uterus, and fallopian tube. M4, Absence of vagina, uterus, fallopian tube, and ovary. (Redrawn from Tarry WR, Duckett JW, Stephens FD. The Mayer-Rokitansky syndrome: Pathogenesis, classification and management. J Urol 1989;136:648–52.)
Bilateral renal agenesis occurs in 1 : 4,800 live births, and has a 3 : 1 male predominance.15 Infants affected with bilateral renal agenesis present with oligohydramnios, pulmonary hypoplasia, Potter’s facies (low-set ears, broad flat nose, a prominent skin fold beginning over the eye and running to the cheek), and the great majority die soon after birth from their pulmonary hypoplasia. The renal arteries and ureters are usually absent, and the bladder is underdeveloped. The vas is usually present, but female genital structures are usually abnormal.16,17 The adrenals are usually present but appear round, instead of flattened, due to the lack of compression by the kidneys.15 Prenatal diagnosis is useful in determining that heroic efforts at extracorporeal membrane oxygenation or hemodialysis are not indicated after delivery.
Supernumerary Kidney
This is a rare condition in which a completely separate kidney is found in addition to two normally positioned kidneys. The additional kidney has its own blood supply and parenchyma, and usually is found caudal to the normal kidney. It is usually smaller than the normally positioned kidney. This additional kidney represents abnormal induction of metanephric blastema by an abnormally directed ureteric bud, either as a separate ureteral bud from the mesonephric duct, or as part of a ‘Y’ duplication. If the supernumerary kidney is located cranial to the normal kidney, the ureter is usually completely separate and may enter the bladder ectopically. Presumably this is a result of a completely separate ureteral bud inducing the metanephric blastema and migrating very low on the mesonephric duct, separate from the normally positioned kidney.18,19 If the ureter ends ectopically, it may present as incontinence in a girl, or as infection in a poorly functioning renal unit. The diagnosis can be difficult.20
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


