Fig. 1.1
The stages of lung development. H&E staining of mouse lungs are shown during the pseudoglandular (a, E13.5), canalicular (b, E15.5), saccular (c, E18.5), and alveolar stages (d–f, postnatal days 2, 8, 21). Progressive expansion of the airways and interstitial thinning are observed (Magnification, 200×)
Table 1.1
The timing and events of pre- and postnatal lung development
Stage | Developmental age | Key morphogenetic events | ||
|---|---|---|---|---|
Human | Mouse | Rat | ||
Embryonic | 4–7 weeks | E9.5–E12 | E11–E13 | Specification and differentiation from the primitive endoderm |
Formation of the lobar bronchi and left/right asymmetry | ||||
Pseudoglandular | 5–17 weeks | E12–E16.5 | E13–E18.5 | Establishment of the prospective conducting (pre-acinar) airways via branching morphogenesis |
Appearance of the presumptive acini—the respiratory units of the lung | ||||
Differentiation of the airway epithelium begins | ||||
Canalicular | 16–26 weeks | E16.5–E17.5 | E18.5–E20 | Establishment of the parenchyma by enhanced capillarization, pneumocyte differentiation, and reduction in mesenchyme |
Surfactant synthesis begins (~22–24 weeks in humans) | ||||
Saccular | 24 weeks–term | E17.5–pnd 4 | E21–pnd 4 | Further development and expansion of the airspaces |
Alveolar | 36 weeks–min. 2 years postnatal | pnd 4–pnd 36 | pnd 4–pnd 60 | Secondary septation dramatically enhances the parenchymal surface area |
Mesenchymal thinning | ||||
Microvascular maturation | ||||
Expression of the definitive distal lung epithelial marker, surfactant protein C (SP-C), is detectable as early as E9.5 in the prospective murine endoderm (Serls et al. 2005). Lung specification and positioning in the foregut relies upon instructive and concentration-dependent signalling by the secreted fibroblast growth factor (FGF) family from the adjacent cardiac mesoderm as well as reciprocal sonic hedgehog (SHH)-GLI signalling between the foregut endoderm and splanchnopleuric mesoderm (Motoyama et al. 1998; Serls et al. 2005). Such tissue interactions at this early stage presage the critical importance that epithelial-mesenchymal signalling will play in the growth and maintenance of the airway epithelium and pulmonary vasculature throughout all stages of lung development.
Right-left asymmetry is already established by 4.5 weeks since the lung anlage is divided into three right-sided and two left-sided diverticula (four right-sided and one left-sided in mice), which corresponds to the adult configuration of the lobar bronchi and lung lobes. Various factors known to be involved in global axis specification have been implicated in this early patterning, including several members of the transforming growth factor (TGF)-β family (Lefty-1 and Lefty-2, Nodal) and Pitx-2, since in all cases, gene deletion in mice causes pulmonary isomerism (Tsukui et al. 1999). Retinoic acid (RA) and SHH appear to be master regulators of restricting appropriate expression of these growth factors (Lin et al. 1999; Meno et al. 1998).
1.1.2 Branching Morphogenesis
The pseudoglandular phase (weeks 5–17 in humans) establishes all the prospective pre-acinar airways (i.e., those proximal to the respiratory bronchioles). This is achieved by branching morphogenesis, in which epithelial buds at the periphery follow a pattern of repetitive dichotomous branching, mediated by reciprocal interactions between the outgrowing epithelium and the surrounding mesenchyme. The importance of such interactions has been demonstrated in classic transplantation experiments using cultured explants, in which the pulmonary epithelium degenerates if isolated from the surrounding mesenchyme, and vice versa (Masters 1976). Conversely, recombination of mesenchyme taken from areas of active branching with non-branching tracheal epithelium induced lung-like epithelial branching, demonstrating both the plasticity of the primordial lung epithelium and the specificity of mesenchymal inductive signalling (Shannon et al. 1998).
Recent analysis in mice has suggested that the orderly development of the millions of branches in the mammalian lung follows a remarkably simple set of only three genetically encoded “subroutines” (Metzger et al. 2008). The first, termed domain branching, sprouts daughter branches at specific sites along and around the circumference of the parent branch. The next two subroutines, planar and orthogonal bifurcation, are responsible for establishing additional tertiary and later generation branches which extend from those developed by domain branching. This occurs from repeated bifurcations of the branch tips along the anterior-posterior axis (planar) or in clusters rotated at 90° (orthogonal). These three subroutines, when stereotyped in just three possible combinations, essentially form all the branching events observed during early pulmonary development. Importantly, these subroutines appear to be under the control of a master routine, which outlines a general developmental plan each branching lineage will follow. In contrast, localized signalling molecules are involved in the direction of the morphogenetic events themselves (mesenchymal invasion, epithelial budding, etc.). These regulators have been the subject of much focus and have been reviewed in detail elsewhere (Cardoso and Lu 2006; Massaro et al. 2004).
1.1.3 Differentiation of the Conducting Airway Epithelium
As one travels along the proximodistal axis of the 223 branches of the mature airways, marked changes in epithelial cell type and distribution are observed, underscoring the variety of regional differences in epithelial function. The pseudostratified and columnar epithelium of the larger airways (branches 20–25) is chiefly composed of basal, ciliated epithelium, goblet cells, and indeterminate cells, all of which rest upon the basal membrane and which overlie layers of smooth muscle cells (Mercer et al. 1994). In addition, submucosal glands are contiguous with the epithelium from the larynx to the small bronchi (Jeffery and Li 1997). Upon reaching the smaller airways (branches 26–223), the epithelial lining decreases in height, becoming principally composed of ciliated cuboidal epithelium and Clara secretory cells. Notably, submucosal glands are absent and goblet cells are normally rare at this level, explaining the absence of mucus secretion (Lumsden et al. 1984).
The diversity and organization of the mature airway epithelium develops as a result of the coordinated differentiation and maturation of the primitive fetal endoderm, beginning in the mid-pseudoglandular stage. Prior to differentiation, the primordial airways are composed of a thick pseudostratified epithelial lining with little to no lumen, resembling the esophagus (Adamson 1997). Such primitive fetal cells have a high nuclear-to-cytoplasmic ratio, indicative of their undifferentiated state, and glycogen, which will fuel later differentiation (Burri 1997). Over the course of airway branching and maturation, this primitive epithelium is dramatically remodelled along the proximodistal axis, beginning in the larger airways and spreading distally, ultimately resulting in airway expansion and decreasing epithelial thickness at all levels of the airways (Adamson 1997). Indeed, airway diameter increases linearly with age in the fetus and neonate, regardless of prematurity (Hislop and Haworth 1989).
Differentiation of both the epithelium and mesenchyme is apparent by the mid-pseudoglandular stage: ciliated epithelium is observed in the tracheobronchial epithelium by the 10th week and in the most peripheral airways by the 13th, while cartilage and smooth muscle cell precursors are found in the trachea by the 10th week, extending towards the segmental bronchi by the 12th week (Bucher and Reid 1961; Burri 1984). During gestation, lung fluid is secreted by the epithelium into the lumen of the future airways. Spontaneous peristaltic contractions of smooth muscle throughout gestation until birth are essential for driving lung fluid distally towards the compliant terminal buds, which may promote airway differentiation and branching via mechanotransduction (Schittny et al. 2000). Submucosal glands are observable by the 14th week, while mucus-containing goblet cells are identifiable by the 13th week in the trachea and large bronchi, extending to the most proximal intrasegmental bronchi by the 24th week (Bucher and Reid 1961). Interestingly, the frequency of goblet cells peaks during mid-gestation, comprising up to 35 % of the bronchial epithelium, before steadily reducing in number to ~12 % in adults (Jeffery 1998; Mercer et al. 1994). Basal cells in the large airways and Clara cells in the bronchioles have been described as early as 10 and 16 weeks gestation, respectively, although the timing of their maturation is speculative and may only fully mature by the mid-canalicular stage (Bucher and Reid 1961; Jeffery 1998; Jeffery et al. 1992).
1.1.4 The Establishment of the Air-Blood Interface
Following branching morphogenesis, the terminal bud epithelium is reduced from a pseudostratified form to simple cuboidal, marking the appearance of the future acini. During the canalicular stage (weeks 16–26), Type II cell differentiation begins, indicated morphologically by the dramatic reduction of intracellular glycogen content, in part from bolstering surfactant synthesis, and the appearance of intracellular lamellar bodies, the storage form of pulmonary surfactant (Adamson 1997). After undergoing structural transformations, such lamellar bodies are secreted into the developing air sacs, where they will form pulmonary surfactant. Importantly, surfactant secretion is present by ~22–24 weeks in the human, and by 80–85 % gestation in most other species, although due to uneven distribution, it appears to be more abundant in apical than basal regions (Burri 1997). Hence, surfactant therapy has been instrumental in the treatment of respiratory distress syndrome (RDS) in premature infants.
The air-blood interface begins to develop as the airspaces widen at the expense of mesenchymal volume and cell number, which is resultant from both mesenchymal apoptosis and a sharp reduction in fibroblast proliferation (Adamson 1997; Stiles et al. 2001). Indeed, during the canalicular stage, the epithelial population doubles in number, reflecting the continuous epithelial cell proliferation and expansion of the airways into the mesenchyme (Adamson 1997). Meanwhile, the developing capillaries begin to “canalize” the lung parenchymal interstitium, forming in close apposition to the epithelium. Concurrently, squamous Type I pneumocytes begin to differentiate from the cuboidal epithelium overlying the invading capillary layers, resulting in a shared basement membrane with the capillary endothelium and thus the formation of the ultrathin air-blood interface alveolar parenchyma. A sharp decline in proliferating epithelium during the canalicular to saccular transition reflects the differentiation of the epithelium into pneumocytes (Adamson 1997). By the end of the saccular phase (26th week–term), the terminal airways exist as primitive saccules which, during the alveolar phase, will develop into functional alveolar units.
Much focus has centered upon the mechanism of Type I cell differentiation, since despite comprising only 8 % of total cells in the adult lung, Type I cells cover over 95 % of the parenchymal surface and are thus at the forefront to the pathogenesis of various major lung diseases (Crystal et al. 2008). Moreover, airway epithelial turnover in the adult is estimated to occur every 30–50 days, although the mechanism of this repopulation is not understood (Crystal et al. 2008). Autoradiographic studies in the rat lung have long suggested that Type I cells differentiate from Type II cells during normal fetal and postnatal development (Kauffman et al. 1974) and after rapid Type II proliferation and replacement of the lung parenchyma following acute lung injury (Evans et al. 1975). However, the stem or progenitor cells which act as the source of new epithelial cells have not been conclusively determined (Crystal et al. 2008). A very recent study using a post-pneumonectomy model of stimulating compensatory lung regrowth has suggested that distinct cells at the bronchoalveolar junction (bronchoalveolar stem cells, BASCs) act as stem cells for a subpopulation of transit-amplifying Type II cells, which in turn differentiate into Type I cells (Nolen-Walston et al. 2008). However, significant cell migration of such BASCs to the sites of airway damage would likely be required, while, based upon the study’s own theoretical estimates, the contribution of BASCs towards alveolar epithelial regeneration may well be minimal.
1.1.5 Development of the Embryonic and Fetal Pulmonary Vascular System
The anatomic origin of the pulmonary circulation is the right ventricle. Throughout gestation, the pulmonary flow is estimated to be only 11 % of the combined biventricular output, but following the closure of the foramen ovale and ductus arteriosus at birth, it receives 100 % of the cardiac output (Mielke and Benda 2001). Patent ductus arteriosus (PDA) in the neonate is associated with deterioration of pulmonary function and alveolar development (i.e., BPD), in part due to pulmonary edema from left-to-right shunt (Massaro et al. 2004). Intriguingly, while pharmacological closure of PDA prevents the incidence of such defects, prophylactic surgical ligation of the ductus arteriosus actually increased the risk of developing BPD, although the mechanism remains unclear (Clyman et al. 2009).
1.1.5.1 The Extrapulmonary Vessels
In the development of the pulmonary vasculature, both the pulmonary trunk and pulmonary arch arteries receive contributions from cardiac neural crest cells, which migrate from the dorsal neural tube and populate the pharyngeal arches and outflow tract of the developing heart (Kirby and Waldo 1995). Between the 4th and 8th weeks of gestation, concurrent with the start of lung budding, the cardiac outflow tract is septated into the aorta and pulmonary trunk from either the fusion of the distal and proximal outflow cushions of the embryonic heart, the elongation of an aortopulmonary septum, or a combination of both paradigms (Webb et al. 2003). During this process, the aortopulmonary septum can be observed growing in between the fourth and sixth paired pharyngeal arches, which will form components of the mature vasculature: the fourth pharyngeal arches contribute to the aortic arch and subclavian arteries, while the right sixth regresses and the left sixth persists to form the ductus arteriosus and the pulmonary arteries by the 7th week (Poelmann and Gittenberger-de Groot 2005).
1.1.5.2 The Intrapulmonary Vessels and Microcirculation
The development of the intrapulmonary vessels (i.e., the branching arterial tree most closely associated with the airways and the pulmonary veins running along the periphery of the terminal respiratory units) and the peripheral microvasculature of the lung parenchyma has sparked much recent investigation (de Mello and Reid 2000; de Mello et al. 1997; Hall et al. 2000, 2002; Maeda et al. 2002; Parera et al. 2005; Schachtner et al. 2000; Schwarz et al. 2009). Although several hypotheses have been put forward, two essential processes are implicated: vasculogenesis, which describes the de novo formation of primitive blood plexuses from the aggregation of mesodermal endothelial progenitor cells (angioblasts), and angiogenesis, wherein new vessels are formed by sprouting from existing ones (Poole and Coffin 1989). Intuitively, one might predict that the two processes follow a successive program. For example, in the establishment of the embryonic vascular network, a primitive vascular plexus formed by vasculogenesis is subsequently remodelled and expanded by angiogenesis (Patan 2000). In the development of the pulmonary vasculature, however, the degree of involvement of either process has not been definitively answered, reflecting differences in methodology and interpretation, and possibly the intrinsic morphological differences among and between human samples and animals models.
The earliest comprehensive studies in the mouse and human described the proximal macrovasculature deriving from angiogenic sprouting of the pulmonary vascular trunks, while the distal microvasculature formed by vasculogenesis of hematopoietic cell lakes diffusely coalescing around actively branching distal airway epithelium, starting in the late embryonic phase (de Mello and Reid 2000; de Mello et al. 1997). In this model, the two processes function concurrently but independently, with fusion of the proximal vasculature and peripheral capillary networks occurring relatively late in pulmonary vascular development (E13–14 in the mouse and 10–11 weeks in the human). In contrast, others described the patency between the proximal and distal vessels—and hence a functional pulmonary circulation—occurring much earlier in development (by E10.5 in mice and 34 days gestation in humans, concurrent with the formation of the first airway branches and tracheal separation from the esophagus) (Hall et al. 2000, 2002; Schachtner et al. 2000). This implicated vasculogenesis as the predominate mechanism of the intrapulmonary vascular system, in which endothelial cell precursors (angioblasts) form primary capillary plexuses surrounding developing epithelial buds in the lung periphery. These plexuses continue to expand and coalesce to form arteries and veins more proximal to the terminal buds, suggesting that unlike the development of the airways, which branch and develop centrifugally from the hilum towards the periphery, the primordial vascular system starts from the periphery and expands proximally (Hall et al. 2000, 2002). However, recent reports have suggested that early pulmonary vascular growth and expansion occurs primarily by angiogenesis (Parera et al. 2005; Schwarz et al. 2009). In agreement with previous studies (Hall et al. 2000; Schachtner et al. 2000), vascular circulation connected to the central vessels appears to be in place at the earliest signs of lung development by the observation of primitive erythrocytes in the lumen of plexiform vessels surrounding growing epithelial buds (at E9.5 in mice) (Parera et al. 2005). However, angiogenesis was suggested to occur predominantly because of the description of these newly formed plexuses remodelling into proximal vessels and the absence of putative angioblasts as assessed by immunostaining for vascular endothelial growth factor receptor (VEGFR)-2 and CD34 (early endothelial markers). Hence, rather than distal vasculogenesis, they proposed distal angiogenesis, in which new capillaries arise from existing ones at the lung periphery (Fig. 1.2). Schwarz and coauthors (2009) agree that the expansion of the pulmonary vasculature in mice occurs primarily by angiogenesis, based upon the co-expression of CD31, phosphorylated VEGFR-2, and fluorescent lectin perfusion, which combined indicated the presence of functional vessels by E11.5. They suggest that previous studies which used VEGFR-2 (rather than phosphorylated VEGFR-2) to indicate angioblasts (and hence vasculogenesis) (Schachtner et al. 2000) reflect uncommitted pluripotent cells. Furthermore, they put forth the interesting model that vessel ingrowth begins within the lung mesenchyme and not in close apposition to the epithelial-mesenchymal interface as is commonly described (e.g., Parera et al. (2005)). In this model, new vessels must then expand and extend from the mesenchyme towards the epithelial-mesenchymal junction via angiogenesis beginning at E14.5, likely as a response to vascular endothelial growth factor (VEGF-A) signalling originating from the basilar epithelium. This would suggest that before E14.5, vessels may not necessarily follow or influence airway branching, although diffusible tissue interactions between the vasculature and epithelium are still required for normal growth and maintenance of both, which is consistent with the degeneration of either tissue layer when cultured in isolation (Gebb and Shannon 2000; Shannon et al. 1998, 1999). Indeed, while chemical inhibition of VEGFR-2 signalling halted vascular growth in E11.5 mouse explants, epithelial branching was only modestly attenuated (Groenman et al. 2007). Hence, disruption of this vessel extension to the primordial parenchyma by inhibiting VEGF-A signalling would prevent the progression of epithelial-endothelial reciprocal signalling and thus the blockage of further lung morphogenesis beyond E14.5, as observed (van Tuyl et al. 2005; Yamamoto et al. 2007).
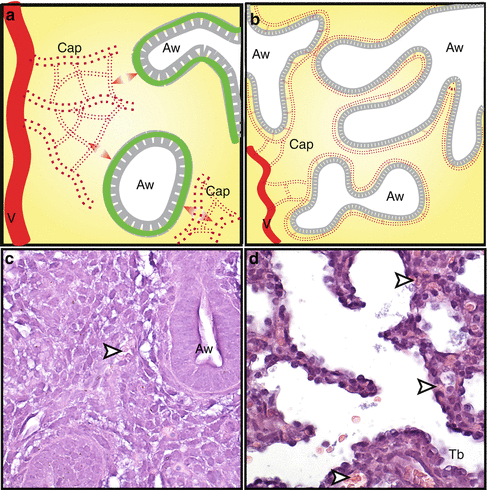
Fig. 1.2
Early vessel formation has been proposed to occur by angiogenic sprouting from preexisting vessels concurrent with the start of lung development. Sprouting occurs within the lung mesenchyme, but not in regions of the epithelial-mesenchymal border early in development (a), although diffusible interactions between the endothelium and epithelium are necessary for continued growth and maintenance (double arrowheads). During the pseudoglandular and canalicular stages, the vasculature expands towards the epithelial-mesenchymal junction, possibly as a response to VEGF-A secretion from the basilar epithelium, forming the air-blood interface (b). H&E staining of mouse lung shows erythrocytes (arrowheads) within the mesenchyme at E13.5 (c) and in close contact with the epithelium at E17.5 (d). Aw developing airway, Cap capillary, Tb terminal bronchiole, V preexisting vessel (Magnification in c and d, 400×)
1.2 Postnatal Lung Development
1.2.1 Postnatal Lung Development: Alveolarization
Normal lung function requires the development of an air-blood barrier of maximal surface area and minimal thinness. This is achieved by the formation of the alveoli beginning in late prenatal human development (week 36) and continuing through early postnatal life in a process termed “alveolarization” (also referred to as “alveologenesis”) (Fig. 1.3). Although birth is often considered the end of organogenesis and the start of growth and maturation, the development and maturation of alveoli in humans and most other mammals is largely, and often exclusively, a postnatal process, ceasing only several years after birth in humans (Burri 1997). The end result of alveolarization and isometric parenchymal growth produces a lung with alveolar and capillary surface areas at least 20 times that of the neonate (Zeltner et al. 1987).
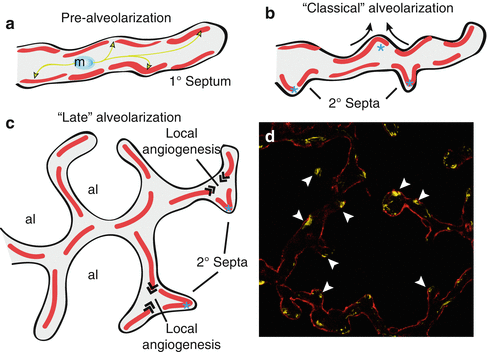
Fig. 1.3
Prior to alveolarization, the future lung alveoli exist as primitive saccules containing a capillary bilayer (a). Myofibroblasts migrate towards distinct sites in the primary saccules and deposit tropoelastin at the future sites of secondary septal formation, ultimately forming localized accumulations of elastin (arrowheads). During classical alveolarization (b), the primary saccules are subdivided into alveoli by secondary septation, while the capillary bilayer is subsequently remodelled into a monolayer during microvascular maturation. During late alveolarization (c), localized angiogenesis at the base of secondary septa is required to maintain the continuity of the capillary monolayer. Secondary septa are observed in an immunostaining with the myofibroblast marker, alpha smooth muscle actin (arrowheads), in a pnd 8 mouse lung doubly stained with the Type I cell marker T1α, which outlines the epithelial surface (d), magnification 400×
1.2.2 The Formation of the Alveoli
Alveolarization occurs as the immature saccules which form the lung parenchyma are subdivided into smaller units by the formation and subsequent extension of secondary septa (singular, “septum”). This is followed by a period of microvascular maturation, in which the double capillary layer lining the alveoli is remodelled into a monolayer by capillary fusion and intussusceptive growth. The seminal reports which introduced this model of alveolarization proposed that the two processes acted somewhat independently of each other: secondary septation was proposed to occur mostly between postnatal days (pnd) 4–14 in the rat, followed successively by microvascular maturation primarily during the third postnatal week (Burri 1974). Hence, the formation of alveoli by further septation was thought impossible following the completion of microvascular maturation, which placed the endpoint of alveolarization in rats and humans conservatively at 3 weeks and 2–3 years postnatal, respectively (Zeltner et al. 1987).
The determination of the endpoint of alveolar development is of much clinical relevance since a longer period of alveolarization increases the window of opportunity for morphological disruptions to occur. For example, alveolar development could arrest without subsequent “catch-up” as a consequence of acute lung disease experienced early in life or from iatrogenic interventions, such as from glucocorticoid therapy in the treatment of asthma. Again, while the postnatal gas-exchange surface area dramatically increases with respect to longitudinal growth (Zeltner et al. 1987), this could arise solely from the isometric growth of the lung parenchyma rather than from the formation of new alveolar septa.
In fact, two recent stereological studies in mice and rats from one group have reported that isometric growth of the lung can only account for a modest increase in septal surface area during postnatal development (Mund et al. 2008; Schittny et al. 2008). By their estimates, the formation of alveoli occurs well beyond the stage of microvascular maturation, ceasing only in young adulthood in mice (pnd 36) and rats (pnd 60). These authors propose that alveolarization proceeds in two phases: “classical” alveolarization occurs prior to microvascular maturation (pnd 4–21) by the formation of secondary septa from immature (capillary bilayer) septa, which accounts for 40 and 50 % of septa present in the adult rat and mouse, respectively, and a second “late” phase (pnd 21–adulthood), in which alveolar septa must arise from mature (capillary monolayer) septa, accounting for 50 and 40 % of septa in the adult rat and mouse, respectively (the remaining 10 % of septa appear to be formed prenatally from branching morphogenesis) (Mund et al. 2008; Schittny et al. 2008). Importantly, the second phase appears to require localized angiogenesis at the base of the newly forming septum, since the capillary microvasculature has already thinned to a monolayer (Mund et al. 2008; Schittny et al. 2008). These results are in agreement with a similar stereological study in rhesus monkeys, which also reported alveolarization continuing through to the 8th postnatal year (Hyde et al. 2007), suggesting that the same may well be true in humans.
While similar analyses in humans are for practical reasons not possible, measurements of pulmonary diffusing capacity for carbon monoxide (DLCO, used indirectly as a measurement of parenchymal surface area) and alveolar volume (V A) in infants aged 2–23 months showed that the DLCO/V A ratio remained constant, suggesting that lung growth occurred primarily by alveolar septation within the first 2 years of life (Balinotti et al. 2009). In contrast, DLCO/V A decreased with increasing age in children ages 6–17 years, consistent with isometric parenchymal lung growth outpacing alveolar septation (Stam et al. 1996). However this does not definitively answer the question of when alveolar development in humans is completed. Indeed, the size of the terminal airspaces in mice and rats was observed to change over the course of postnatal development as a result of the changing rates of septal formation and parenchymal growth, which vary over maturation, although with some interspecies variability (Mund et al. 2008; Schittny et al. 2008). Hence, whether alveolarization in humans principally occurs en masse in a relatively short period of time (so-called bulk alveolarization), or more gradually, and perhaps variably, alongside longitudinal growth, has yet to be determined.
1.2.3 The Regulation of Secondary Septation
BPD is a condition which affects nearly one third of neonates with birth weights less than 1,000 g (Walsh et al. 2006). The aetiology of the disease is multifactorial and develops in response to lung injury experienced from both environmental factors (hyperoxia relative to in utero, infection, nutritional deficits, lung immaturity) and strategies designed to manage premature infant care (mechanical ventilation, antenatal steroid administration). Genetic susceptibility, such as with respect to surfactant synthesis and inflammatory response, is also being recognized as a significant contributing factor (Abman et al. 2008).
BPD is presently characterized by an arrest in alveolar septation and dysmorphic vascular growth, leading to a reduction in gas-exchange surface area and respiratory potential (Bourbon et al. 2005). Similarly, gas exchange is impaired in adult-onset emphysema, which arises from damage to preexisting alveoli (Morris and Sheppard 2006). Studies which identify the fundamental regulators which orchestrate normal alveolarization and how these pathways are disrupted in conditions such as BPD and emphysema are critical for the development of therapies which can treat these debilitating diseases. However, the mechanisms involved in the various morphogenetic processes of alveolarization are not well understood. Multiple stimuli, including growth and transcription factors, extracellular matrix (ECM) proteins, hormones, and oxygen exposure coordinated through autocrine and paracrine interactions between the epithelium, fibroblast derivatives, and the ECM, work together in concert to sculpt the alveolar architecture (Roth-Kleiner and Post 2005). Conversely, dysregulation of these interactions results in arrested alveolar development. Next we provide a short updated review of factors known to be essential for normal alveolarization and which have largely been uncovered in the context of chronic lung diseases, such as BPD.
1.2.3.1 ECM
The ECM comprises the basal lamina and interstitium subjacent to the parenchyma, and its composition is critical for the mechanical properties of the lung as well as the regulation of cell growth, morphogenesis, and differentiation. Although the principal component of the ECM is collagen and adherent glycoproteins, elastic fibers interwoven into the structure at the septal interstitial tips and alveolar free edges contribute most to the structural integrity and compliance of the lung and stretch-induced signal transduction during each respiratory cycle (Mercer and Crapo 1990). Elastic fibers are formed when tropoelastin, secreted from myofibroblasts, coalesces and cross-links upon a microfibril scaffolding. The attachment of the scaffolding to the cell membrane is regulated by various matrix proteins, while cross-linking is induced by the action of lysyl oxidases, thus producing insoluble elastin fibers (Bland et al. 2008). The absolute quantity of parenchymal elastic tissue increases slowly during gestational weeks 22–30, but rapidly increases thereafter, correlating with its proposed function in secondary crest formation (Thibeault et al. 2000).
Prior to secondary crest eruption from the saccular walls, myofibroblasts proliferate and migrate to discrete areas of the saccular walls, where they deposit tropoelastin at the sites of future secondary crest formation, thus forming the elastic fibers. It has been suggested that septal formation is initiated by mechanical stress applied to these elastic fibers which lift up the alveolar walls, although the mechanism is unclear (Burri 1997). Indeed, tenascin-C, known to be expressed in relation to increasing mechanical load, is present at the rising septal tips (Massaro et al. 2004). Destruction of elastin fibers contributes to the pathogenesis of emphysema, while elastin-null mice are perinatally lethal and exhibit marked reduction in tissue septa and distal air sacs, reminiscent of emphysema (Wendel et al. 2000). Furthermore, mice with only one elastin-encoding allele have increased susceptibility to smoke-induced emphysema (Shifren et al. 2007).
Interestingly, dysregulation of elastin deposition has been associated with alveolar malformation. Ovine and murine mechanical ventilation models of BPD as well as BPD infants display abnormally enhanced elastin deposition and distribution throughout the alveolar airspaces and secondary crests instead of focal concentrations at the septal tips (Bland et al. 2008; Pierce et al. 1997; Thibeault et al. 2000). In these models, lung expression of growth factors known to regulate lung septation, such as VEGF-A, platelet-derived growth factor alpha (PDGF-A), and their associated receptors, was downregulated, while factors known to enhance elastin production (TGF-α and TGF-β1) were upregulated (Bland et al. 2008). Tropoelastin deposition is known to occur in response to mechanical stretch and hyperoxia, although curiously, the expression of elastin-regulating matrix proteins was not correspondingly increased. It is possible that enhanced tropoelastin expression and increased serine elastase activity in response to these stressors result in abundant but disorganized elastic fibers due to the absence of a concomitant increase in elastin-regulating proteins, ultimately leading to alveolar malformation (Bland et al. 2008).
Two essential modulator groups of ECM turnover are the matrix metalloproteinases (MMPs) and their natural regulators, tissue inhibitor of metalloproteinases (TIMPs). Presently, 24 MMPs exist in humans, many of which are differentially expressed during the stages of lung development, although the significance of these patterns of expression has yet to be fully elucidated (Greenlee et al. 2007). They are capable of degrading a variety of targets, particularly ECM molecules such as collagen, which is indicative of their role in ECM remodelling during normal development and injury-induced repair (Greenlee et al. 2007). For example, the expression of MMP-2 and MMP-14 increases substantially at birth, while deletion of these genes is associated with abnormal alveolar development (Atkinson et al. 2005; Kheradmand et al. 2002). Conversely, altered levels of MMP and TIMP expression, as well as altered expression of other proteases, such as cathepsins, are a hallmark of inflammatory diseases, such as BPD (Altiok et al. 2006; Ambalavanan et al. 2008; Chetty et al. 2008; Schulz et al. 2004). Finally, with respect to the emerging concept of BPD involving a genetic predisposition (Abman et al. 2008), single-nucleotide polymorphisms of MMP-16 have recently been implicated in the protection against the development of BPD, although curiously, MMP-16 was elevated in a rat model of BPD (Hadchouel et al. 2008).
1.2.3.2 Growth Factors
The sites of secondary crest formation appear to be nonrandom, but instead arise from the deposition of elastin into the alveolar walls by myofibroblasts. One factor critical to the appropriate migration of myofibroblasts to the sites of secondary crest formation is PDGF-A (Bostrom et al. 1996; Lindahl et al. 1997). Myofibroblasts, or their precursors, respond to PDGF-A chemoattractant secretions from the epithelium by migrating to the peripheral sites of the lung and depositing elastin. Deletion of PDGF-A results in a dramatic reduction in elastin synthesis and consequently the absence of secondary crest formation (Lindahl et al. 1997). It has been proposed that PDGF-A expression is finely regulated by morphogen gradients which precisely localize migrating myofibroblasts to appropriate sites of septal initiation (Prodhan and Kinane 2002). Although the identity of the morphogen involved has not been identified, candidate molecules include SHH and RA (Prodhan and Kinane 2002).
Critical roles for FGFs and their receptors, FGFR-1–5, have been demonstrated in both prenatal and postnatal lung development. During branching morphogenesis, FGF-7 and FGF-10 are expressed in the mesenchyme at areas of active branching, while FGFR-2 is complementarily expressed in the epithelium (Bellusci et al. 1997; Post et al. 1996). Furthermore, disruption of FGF-7, FGF-10, or FGFR-2 signalling through targeted deletion, antisense knockdown, or neutralizing antibodies results in diminished or absent branching, implicating the importance of reciprocal epithelial-mesenchymal interactions during morphogenesis (De Moerlooze et al. 2000; Post et al. 1996; Sekine et al. 1999; Shiratori et al. 1996). Interestingly, dominant negative expression of FGFR-2 during embryonic, but not postnatal, lung development results in irreversible emphysema, which demonstrates how alveolarization can be indirectly impacted by dysregulation of proliferation or differentiation of precursors during the fetal period (Hokuto et al. 2003).
FGF signalling is also directly required for septation during alveolarization: double knockout of FGFR-3 and FGFR-4 results in exuberant elastin deposition and consequently a failure of secondary septa to form, demonstrating the importance of interactions between soluble growth factors and the ECM (Weinstein et al. 1998). FGF-7 and FGF-2 are upregulated following hyperoxia-induced alveolar damage and may play a role in inducing reparative Type II to Type I cell transition (Buch et al. 1995). Furthermore, inhibition of FGF-2 signalling through FGFR-1 prevents normal postnatal rat lung cell apoptosis, while FGF-7 disruption reduces alveolar number (Padela et al. 2008; Yi et al. 2006).
FGF-7 is also an important stimulator of proliferation and maturation of developing Type II pneumocytes (Chelly et al. 1999) and is a positive predictor for absence of BPD in premature infants (Danan et al. 2002). FGF-7 also protects against injury caused by hyperoxia (Frank 2003) and/or mechanical ventilation (Welsh et al. 2000). Interestingly, although FGF-7 did not prevent attenuated septation associated with hyperoxia (Welsh et al. 2000), exogenous FGF-7 stimulated compensatory lung growth following pneumonectomy by enhancing alveolar cell proliferation (Kaza et al. 2002). Finally, neonatal mice exposed to hyperoxic conditions during alveolarization display reduced FGFR-3, FGFR-4, and FGF-7 expression (Park et al. 2007).
Although TGF-β has received much attention for its function as a negative regulator of branching morphogenesis, it has more recently been implicated as an important factor in both alveolar formation and architectural maintenance (Alejandre-Alcazar et al. 2008). TGF-β suppresses Type II epithelial cell differentiation into Type I pneumocytes in human fetal cell culture, while it is downregulated upon glucocorticoid-induced differentiation (McDevitt et al. 2007). Interestingly, following an acute lung injury to Type I cells, Type II cells undergo phases of proliferation and differentiation, which are paradoxically suppressed and enhanced, respectively, by TGF-β (Bhaskaran et al. 2007). Disruption of TGF-β signalling resulted in emphysema-like airspace enlargement due to an abundance of matrix metalloproteinases, while conversely, introduction of excessive TGF-β led to pulmonary fibrosis, likewise indicating the importance of TGF-β regulation of ECM homeostasis (Bonniaud et al. 2004). TGF-β is also increased in the airway secretions of preterm infants with BPD (Kotecha et al. 1996). Moreover, enhanced TGF-β signalling and dysregulation of ECM-remodelling proteins are associated with hyperoxia-induced BPD in neonatal mouse models (Alejandre-Alcazar et al. 2007), while prenatal administration of a TGF-β-neutralizing antibody partially rescued the BPD phenotype (Nakanishi et al. 2007).
1.2.3.3 Transcription Factors
Despite the importance of transcriptional programs which mediate lung morphogenesis, transcription factors which regulate alveolarization specifically are not well described. Interestingly, many transcription factors which are critical for fetal lung development are downregulated during alveolarization. For example, GATA-6, despite being important for branching morphogenesis, is not expressed postnatally, although postnatal GATA-6 overexpression in transgenic mice results in impaired alveolarization (Liu et al. 2003). Similarly, thyroid transcription factor-1 (TTF-1) is expressed in the epithelium of the fetal lung buds but becomes restricted to Type II cells postnatally (Wert et al. 2002). TTF-1 overexpression decreased alveolar septation and caused severe chronic pulmonary inflammation and Type II cell hyperplasia (Wert et al. 2002). In both cases, GATA-6 and TTF-1 appear to affect Type II to Type I cell differentiation with resultant impaired alveolarization if misexpressed.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


