Chapter 80 Defects in Metabolism of Lipids
80.1 Disorders of Mitochondrial Fatty Acid β-Oxidation
Genetic defects have been identified in nearly all of the known steps in the fatty acid oxidation pathway; all are recessively inherited (Table 80-1).
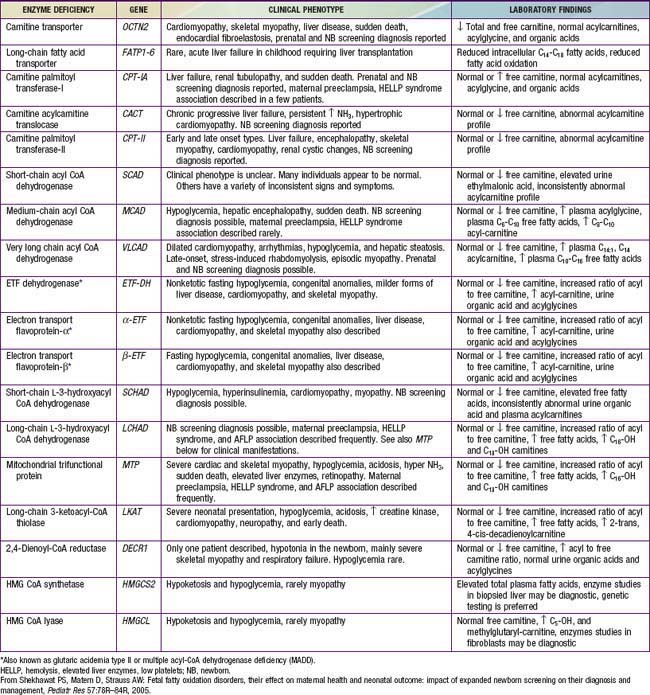
Figures 80-1 and 80-2 outline the steps involved in the oxidation of a typical long-chain fatty acid. In the carnitine cycle, fatty acids are transported across the barrier of the inner mitochondrial membrane as acylcarnitine esters. Within the mitochondria, successive turns of the 4-step β-oxidation cycle convert the coenzyme A (CoA)-activated fatty acid to acetyl CoA units. Two to three different chain-length specific isoenzymes are needed for each of these β-oxidation steps to accommodate the different-sized fatty acyl CoA species. The electron transfer pathway carries electrons generated in the 1st β-oxidation step (acyl CoA dehydrogenase) to the electron transport chain for adenosine triphosphate (ATP) production, while electrons generated from the third step (3-hydroxyacyl CoA dehydrogenase) enter the respiratory chain at the level of complex 1. Most of the acetyl CoA generated from hepatic β-oxidation flows through the pathway of ketogenesis to form β-hydroxybutyrate and acetoacetate.
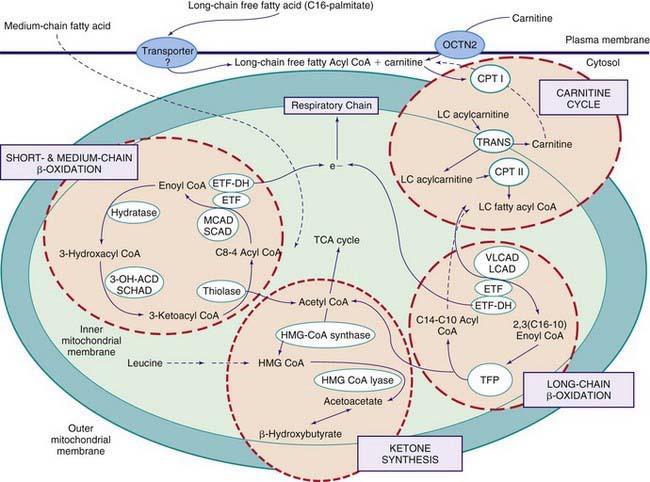
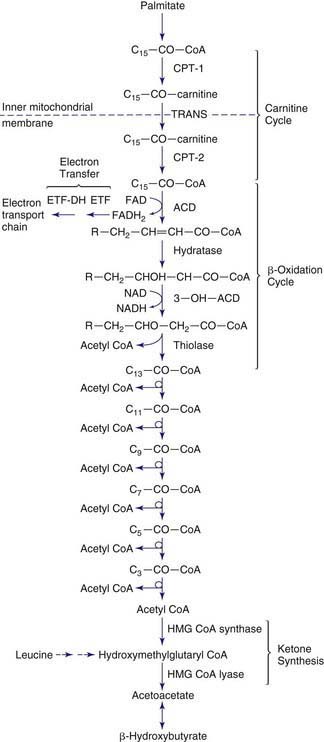
Defects in the β-Oxidation Cycle
Medium-Chain Acyl CoA Dehydrogenase (MCAD) Deficiency
Treatment
Acute illnesses should be promptly treated with intravenous fluids containing 10% dextrose to treat or prevent hypoglycemia and to suppress lipolysis as rapidly as possible (Chapter 86). Chronic therapy consists of avoiding fasting. This usually requires simply adjusting the diet to ensure that overnight fasting periods are limited to <10-12 hr. Restricting dietary fat or treatment with carnitine is controversial. The necessity for active therapeutic intervention for individuals with the T199C variant has not yet been established.
Long-Chain 3-Hydroxyacyl CoA Dehydrogenase (LCHAD)/Mitochondrial Trifunctional Protein (TFP) Deficiency
Defects in the Carnitine Cycle
Defects in Electron Transfer Pathway
Defects in Ketone Synthesis Pathway
Defects in Ketone Utilization
De Leon DD, Stanley CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3:57-68.
Gillingham MB, Weleber RG, Neuringer M, et al. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005;86:124-133.
Greenberg CR, Dilling LA, Thompson GR, et al. The paradox of the carnitine palmitoyltransferase type 1a P479L variant in Canadian Aboriginal populations. Mol Genet Metab. 2009;96:201-207.
Hsu HW, Zytkovicz TH, Comeau AM, et al. Spectrum of medium-chain acyl-CoA dehydrogenase deficiency detected by newborn screening. Pediatrics. 2008;121:e1108-e1114.
Jethva R, Bennett MJ, Vockley J. Short-chain acyl-coenzyme A dehydrogenase deficiency. Mol Genet Metab. 2008;95:195-200.
Longo N, di San Filippo CA, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C:77-85.
Loughrey C, Bennett MJ. Screening for MCAD deficiency in newborns. BMJ. 2009;338:843-846.
Molven A, Matre GE, Duran M, et al. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes. 2004;53:221-227.
Schulze A, Matern D, Hoffmann GF. Newborn Screening. In: Sarafoglou K, Hoffmann GF, Roth KS, editors. Pediatric endocrinology and inborn errors of metabolism. New York: McGraw-Hill; 2009:17-32.
Shekhawat PS, Matern D, Strauss AW. Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management. Pediatr Res. 2005;57:78R-86R.
Strauss AW, Andresen BS, Bennett MJ. Mitochondrial fatty acid oxidation defects. In: Sarafoglou K, Hoffmann GF, Roth KS, editors. Pediatric endocrinology and inborn errors of metabolism. New York: McGraw-Hill; 2009:51-70.
van Maldegem BT, Duran M, Wanders RJA, et al. Fasting and fat-loading tests provide pathophysiological insight into short-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 2010;156:121-127.
Wilcken B, Haas M, Joy P, et al. Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet. 2007;369:37-42.
80.2 Disorders of Very Long Chain Fatty Acids
Peroxisomal Disorders
Etiology
Peroxisomal disorders are subdivided into 2 major categories (Table 80-2).
Table 80-2 CLASSIFICATION OF PEROXISOMAL DISORDERS
A: DISORDERS OF PEROXISOME IMPORT
A1: Zellweger syndrome
A2: Neonatal adrenoleukodystrophy
A3: Infantile Refsum disease
A4: Rhizomelic chondrodysplasia punctata
B: DEFECTS OF SINGLE PEROXISOMAL ENZYME
B1: X-linked adrenoleukodystrophy
B2: Acyl CoA oxidase deficiency
B3: Bifunctional enzyme deficiency
B4: Peroxisomal thiolase deficiency
B5: Classic Refsum disease
B6: 2-Methylacyl CoA racemase deficiency
B7: DHAP acyltransferase deficiency
B8: Alkyl-DHAP synthase deficiency
B9: Mevalonic aciduria
B10: Glutaric aciduria type III
B11: Hyperoxaluria type I
B12: Acatalasemia
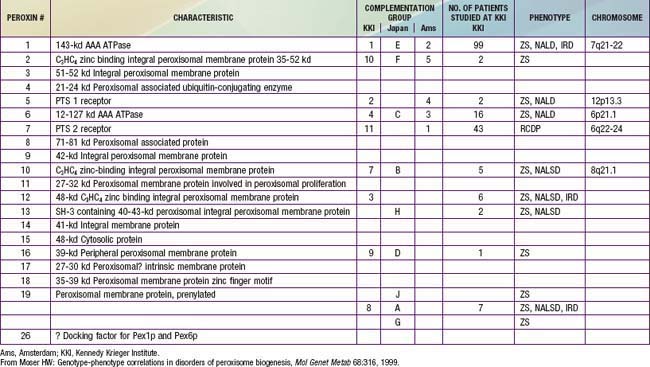
In category A, the peroxisomal biogenesis disorders (PBD), the basic defect is the failure to import one or more proteins into the organelle. In category B, defects affect a single peroxisomal protein. The peroxisome is present in all cells except mature erythrocytes and is a subcellular organelle surrounded by a single membrane; >50 peroxisomal enzymes are identified. Some enzymes are involved in the production and decomposition of hydrogen peroxide; others are concerned with lipid and amino acid metabolism. Most peroxisomal enzymes are first synthesized in their mature form on free polyribosomes and enter the cytoplasm. Proteins that are destined for the peroxisome contain specific peroxisome targeting sequences (PTS). Most peroxisomal matrix proteins contain PTS1, a 3-amino acid sequence at the carboxyl terminus. PTS2 is an amino-terminal sequence that is critical for the import of enzymes involved in plasmalogen and branched-chain fatty acid metabolism. Import of proteins involves a complex series of reactions that involves at least 23 distinct proteins. These proteins are referred to as peroxins encoded by PEX genes. Table 80-3 summarizes the PEX genes that are defective in human disease states.
Epidemiology
Except for X-linked adrenoleukodystrophy (X-ALD), all the peroxisomal disorders in Table 80-2 are autosomal recessive traits. X-ALD is the most common peroxisomal disorder, with an estimated incidence of 1/17,000. The combined incidence of the other peroxisomal disorders is estimated to be 1/50,000.
Pathogenesis
It is likely that all pathologic changes are secondary to the peroxisome defect. Multiple peroxisomal enzymes fail to function in the PBD (Table 80-4). The enzymes that are diminished or absent are synthesized but are degraded abnormally fast because they may be unprotected outside of the peroxisome. It is not clear how defective peroxisome functions lead to the widespread pathologic manifestations.
Table 80-4 ABNORMAL LABORATORY FINDINGS COMMON TO DISORDERS OF PEROXISOME BIOGENESIS
Peroxisomes absent to reduced in number
Catalase in cytosol
Deficient synthesis and reduced tissue levels of plasmalogens
Defective oxidation and abnormal accumulation of very long chain fatty acids
Deficient oxidation and age-dependent accumulation of phytanic acid
Defects in certain steps of bile acid formation and accumulation of bile acid intermediates
Defects in oxidation and accumulation of L-pipecolic acid
Increased urinary excretion of dicarboxylic acids
The PBD are associated with genetically determined import defects. The PBD have been subdivided into 12 complementation groups. The molecular defects have been defined in 10 of these groups (see Table 80-3). The pattern and severity of pathologic features vary with the nature of the import defects and the degree to which import is impaired. These gene defects lead to disorders that were named before their relationship to the peroxisome was recognized, namely, Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and rhizomelic chondrodysplasia punctata (RCDP). The first 3 disorders are considered to form a clinical continuum, with ZS the most severe, IRD the least severe, and NALD intermediate. They can be caused by 11 different gene defects, which involve mainly the import of proteins that contain the PTS1 targeting signal; the gene defects cannot be distinguished on the basis of clinical features. The clinical severity varies with the degree to which protein import is impaired. Mutations that abolish import completely are often associated with the ZS phenotype, whereas a missense mutation, in which some degree of import function is retained, leads to the somewhat milder phenotypes. A defect in PEX7, which involves the import of proteins that utilize PTS2, is associated with RCDP. PEX7 defects that leave import partially intact are associated with milder phenotypes, some of which resemble classic Refsum disease.
PBD With Milder or Atypical Phenotypes
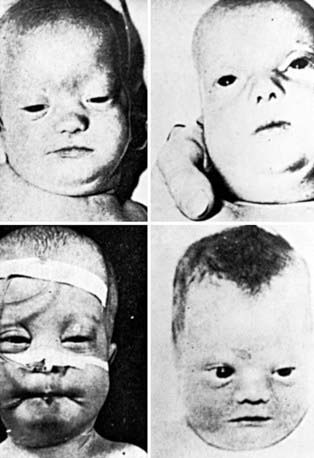
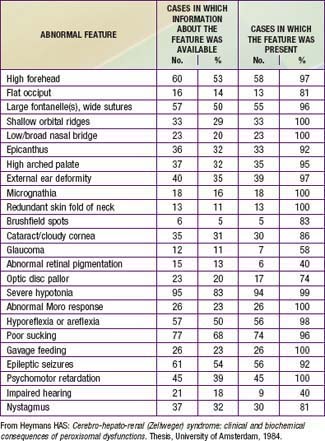
Newborn infants with Zellweger syndrome show striking and consistent, recognizable abnormalities. Of central diagnostic importance are the typical facial appearance (high forehead, unslanting palpebral fissures, hypoplastic supraorbital ridges, and epicanthal folds; Fig. 80-3), severe weakness and hypotonia, neonatal seizures, and eye abnormalities (cataracts, glaucoma, corneal clouding, Brushfield spots, pigmentary retinopathy, and nerve dysplasia). Because of the hypotonia and “mongoloid” appearance, Down syndrome may be suspected. Infants with Zellweger syndrome rarely live more than a few months. More than 90% show postnatal growth failure. Table 80-5 lists the main clinical abnormalities.
Rhizomelic Chondrodysplasia Punctata (Rcdp)
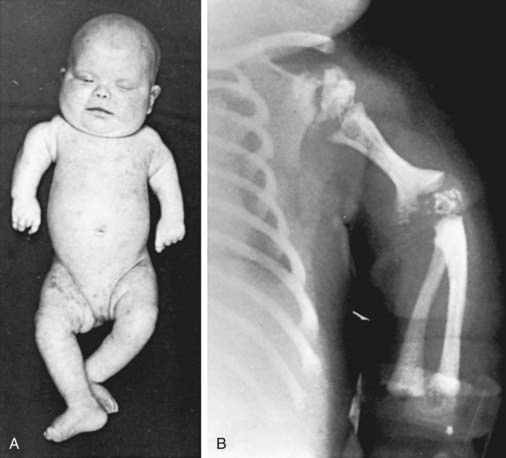
This disorder is characterized by the presence of stippled foci of calcification within the hyaline cartilage and is associated with dwarfing, cataracts (72%), and multiple malformations due to contractures. Vertebral bodies have a coronal cleft filled by cartilage that is a result of an embryonic arrest. Disproportionate short stature affects the proximal parts of the extremities (Fig. 80-4A). Radiologic abnormalities consist of shortening of the proximal limb bones, metaphyseal cupping, and disturbed ossification (Fig. 80-4B). Height, weight, and head circumference are less than the 3rd percentile, and these children are severely retarded mentally. Skin changes such as those observed in ichthyosiform erythroderma are present in about 25% of patients.
Isolated Defects of Peroxisomal Fatty Acid Oxidation
The disorders labeled B1 through B3 (see Table 80-2) each involve 1 of 3 enzymes involved in peroxisomal fatty acid oxidation. Their clinical manifestations resemble those of the Zellweger syndrome/neonatal ALD/infantile Refsum disease continuum; they can be distinguished from disorders of peroxisome biogenesis by laboratory tests. Defects of bifunctional enzyme are common and are found in about 15% of patients with the Zellweger syndrome/neonatal ALD/infantile Refsum disease phenotype. Patients with isolated acyl CoA oxidase deficiency have a somewhat milder phenotype that resembles that of neonatal ALD.
Isolated Defects of Plasmalogen Synthesis
Plasmalogens are lipids in which the 1st carbon of glycerol is linked to an alcohol rather than a fatty acid. They are synthesized through a complex series of reactions, the 1st two steps of which are catalyzed by the peroxisomal enzymes dihydroxyacetone phosphate alkyl transferase and synthase. Deficiency of either of these enzymes (B4 and B5 in Table 80-2) leads to a phenotype that is clinically indistinguishable from the peroxisomal import disorder RCDP. This latter disorder is caused by a defect in PEX7, the receptor for peroxisome targeting sequence 2. It shares the severe deficiency of plasmalogens with disorders B4 and B5 but, in addition, has defects of phytanic oxidation. The fact that disorders B4 and B5 are associated with the full phenotype of RCDP suggests that a deficiency of plasmalogens is sufficient to produce it.
Laboratory Findings
Laboratory tests for peroxisomal disorders can be viewed at three levels of complexity.
Level 1: Does the Patient have a Peroxisomal Disorder?
This can be resolved by noninvasive tests that are generally available (Table 80-6). Measurement of plasma VLCFA is the most commonly used assay. Whereas plasma VLCFA levels are elevated in many patients with peroxisomal disorders, this is not always the case. The most important exceptions are RCDP, in which VLCFA levels are normal, but plasma phytanic acid levels are increased and red blood cell plasmalogen levels are reduced. In some other peroxisomal disorders, the biochemical abnormalities are still more restricted. Therefore, a panel of tests is recommended and includes plasma levels of VLCFA and phytanic, pristanic, and pipecolic acids and red blood cell levels of plasmalogens. Tandem mass spectrometry techniques also permit convenient quantitation of bile acids in plasma and urine. This panel of tests can be performed on 2 mL samples of venous blood and permits detection of most peroxisomal disorders. Furthermore, normal results make the presence of a peroxisomal disorder unlikely.
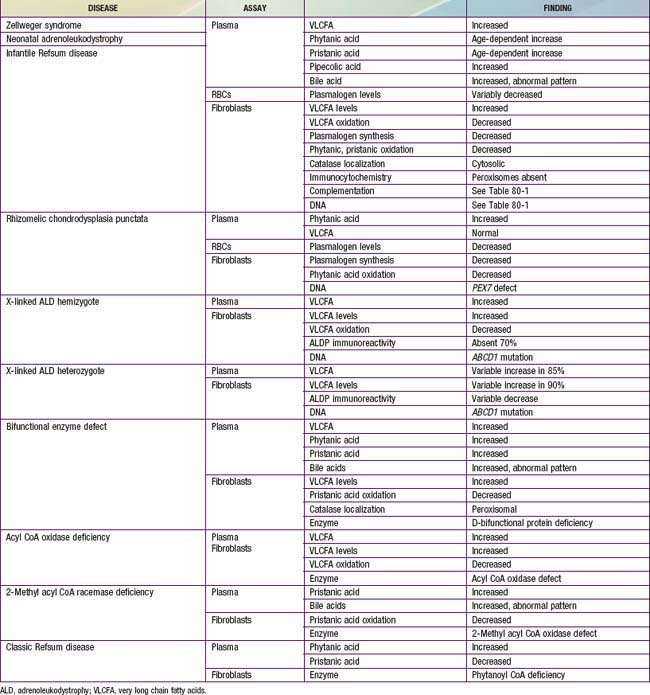
Level 2: What is the Precise Nature of the Peroxisomal Disorder?
Table 80-6 lists the main biochemical abnormalities in the various peroxisomal disorders. When combined with the clinical presentation, the panel of level 1 tests (see earlier) is often sufficient to identify the precise nature of the defect. Elevated plasma VLCFA levels permit the precise diagnosis of X-ALD in male patients. Marked reduction of erythrocyte plasmalogen levels combined with elevated plasma phytanic acid permits precise diagnosis in a patient with the clinical features of RCDP. Classic Refsum disease can be diagnosed by demonstration of increased plasma phytanic acid combined with normal or reduced levels of pristanic acid levels, while in D-bifunctional enzyme deficiency and 2-methylacyl CoA racemase deficiency, the levels of pristanic and phytanic acid are both increased. Precise identification of some peroxisomal disorders may require more extensive studies in cultured skin fibroblasts. This may be required for the differentiation of PBD from defects in bifunctional enzyme. In PBD, the patient’s peroxisomes are absent and catalase is in the soluble fraction, whereas in bifunctional enzyme defect, peroxisomes are present and catalase is in the particulate fraction. Fibroblast studies are required to identify the nature of the molecular defect in PBD. Whether such specialized studies are clinically warranted depends on individual circumstances. Precise definition of the defect in a proband may improve the precision of prenatal diagnosis in at-risk pregnancies, and it is required for carrier detection. It is also of value in setting prognosis. Precise characterization is of prognostic value in patients with PEX1 defects. This defect is present in approximately 60% of PBD patients, and about half of the PEX1 defects have the G843D allele, which is associated with a significantly milder phenotype than is found in other mutations.
Diagnosis
There are several noninvasive laboratory tests that permit precise and early diagnosis of peroxisomal disorders (see Table 80-6). The challenge in PBD is to differentiate them from the large variety of other conditions that can cause hypotonia, seizures, failure to thrive, or dysmorphic features. Experienced clinicians can readily recognize classic Zellweger syndrome by its clinical manifestations. PBD patients often do not show the full clinical spectrum of disease and may be identifiable only by laboratory assays. Clinical features that may serve as indications for these diagnostic assays include severe psychomotor retardation; weakness and hypotonia; dysmorphic features; neonatal seizures; retinopathy, glaucoma, or cataracts; hearing deficits; enlarged liver and impaired liver function; and chondrodysplasia punctata. The presence of one or more of these abnormalities increases the likelihood of this diagnosis. Atypical milder forms presenting as peripheral neuropathy have also been described.
Genetic Counseling
All the peroxisomal disorders, except hyperoxaluria type 1, can be diagnosed prenatally in the 1st or 2nd trimester. The tests are similar to those described for postnatal diagnosis (see Table 80-6) and use chorionic villus sampling or amniocytes. More than 300 pregnancies have been monitored, and more than 60 affected fetuses have been identified without diagnostic error. Because of the 25% recurrence risk, couples with an affected child must be advised about the availability of prenatal diagnosis. Heterozygotes can be identified in X-ALD and in those disorders in which the molecular defect has been identified (see Table 80-3).
Adrenoleukodystrophy (X-Linked)
Etiology
The key biochemical abnormality is the tissue accumulation of unbranched saturated VLCFA, with a carbon chain length of 24 or more. Excess hexacosanoic acid (C26 : 0) is the most striking and characteristic feature. This accumulation of fatty acids is caused by genetically deficient peroxisomal degradation of fatty acid. The key biochemical defect involves the impaired function of peroxisomal lignoceroyl CoA ligase, the enzyme that catalyzes the formation of the CoA derivative of VLCFA. The gene that is defective (ABCD1) codes for a peroxisomal membrane (ALDP). More than 400 distinct mutations have been identified, and most families have a mutation that is “private” (unique to that kindred) and are updated on the website, www.x-ald.nl. The gene has been mapped to chromosome Xq28. The mechanism by which the ALDP defect leads to VLCFA accumulation and the pathology of X-ALD is unknown.
Laboratory and Radiographic Findings
CT And MRI
Patients with childhood cerebral or adolescent ALD show cerebral white matter lesions that are characteristic with respect to location and attenuation patterns on MRI. In 80% of patients, the lesions are symmetric and involve the periventricular white matter in the posterior parietal and occipital lobes. About 50% show location of a garland of accumulated contrast material adjacent and anterior to the posterior hypodense lesions (Fig. 80-5A). This zone corresponds to the zones of intense perivascular lymphocytic infiltration where the blood-brain barrier breaks down. In 12% of patients, the initial lesions are frontal. Unilateral lesions that produce a mass effect suggestive of a brain tumor may occur. MRI provides a clearer delineation of normal and abnormal white matter than does CT and may demonstrate abnormalities missed by CT (Fig. 80-5B).
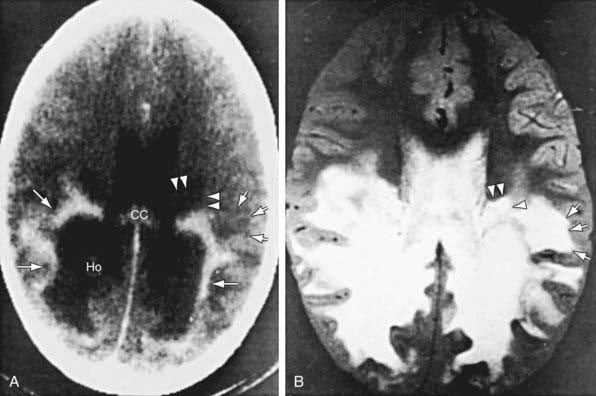
Diagnosis and Differential Diagnosis
The earliest manifestations of childhood cerebral ALD are difficult to distinguish from the more common attention-deficit disorders or learning disabilities. Rapid progression, signs of dementia, or difficulty in auditory discrimination suggest ALD. Even in early stages, CT or MRI may show strikingly abnormal changes. Other leukodystrophies (Chapters 592 and 605.10) or multiple sclerosis (Chapter 593.1) may mimic these radiographic findings. Definitive diagnosis depends on demonstration of VLCFA excess, which occurs only in X-ALD and the other peroxisomal disorders. The latter may be distinguished from X-ALD by their clinical presentation during the neonatal period.
Treatment
Corticosteroid replacement for adrenal insufficiency or adrenocortical hypofunction is effective (Chapter 569). It may be lifesaving and increase general strength and well-being, but it does not alter the course of the neurologic disability.



