Chapter 81 Defects in Metabolism of Carbohydrates
Carbohydrate synthesis and degradation provide the energy required for most metabolic processes. The important carbohydrates include 3 monosaccharides—glucose, galactose, and fructose—and a polysaccharide, glycogen. The relevant biochemical pathways of these carbohydrates are shown in Figure 81-1. Glucose is the principal substrate of energy metabolism. A continuous source of glucose from dietary intake, gluconeogenesis, and glycogenolysis of glycogen maintains normal blood glucose levels. Metabolism of glucose generates adenosine triphosphate (ATP) via glycolysis (conversion of glucose or glycogen to pyruvate), mitochondrial oxidative phosphorylation (conversion of pyruvate to carbon dioxide and water), or both. Dietary sources of glucose come from ingesting polysaccharides, primarily starch and disaccharides, including lactose, maltose, and sucrose. Oral intake of glucose is intermittent and unreliable. Glucose made de novo from amino acids, primarily alanine (gluconeogenesis), contributes to maintaining the euglycemic state, but this process requires time. The breakdown of hepatic glycogen provides the rapid release of glucose, which maintains a constant blood glucose concentration. Glycogen is also the primary stored energy source in muscle, providing glucose for muscle activity during exercise. Galactose and fructose are monosaccharides that provide fuel for cellular metabolism; their role is less significant than that of glucose. Galactose is derived from lactose (galactose + glucose), which is found in milk and milk products. Galactose is an important energy source in infants, but it is 1st metabolized to glucose. Galactose (exogenous or endogenously synthesized from glucose) is also an important component of certain glycolipids, glycoproteins, and glycosaminoglycans. The dietary sources of fructose are sucrose (fructose + glucose, sorbitol) and fructose itself, which is found in fruits, vegetables, and honey.
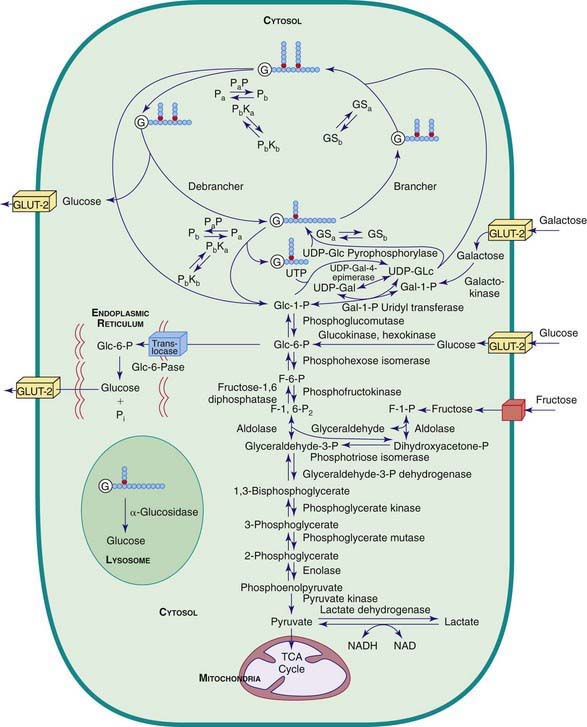
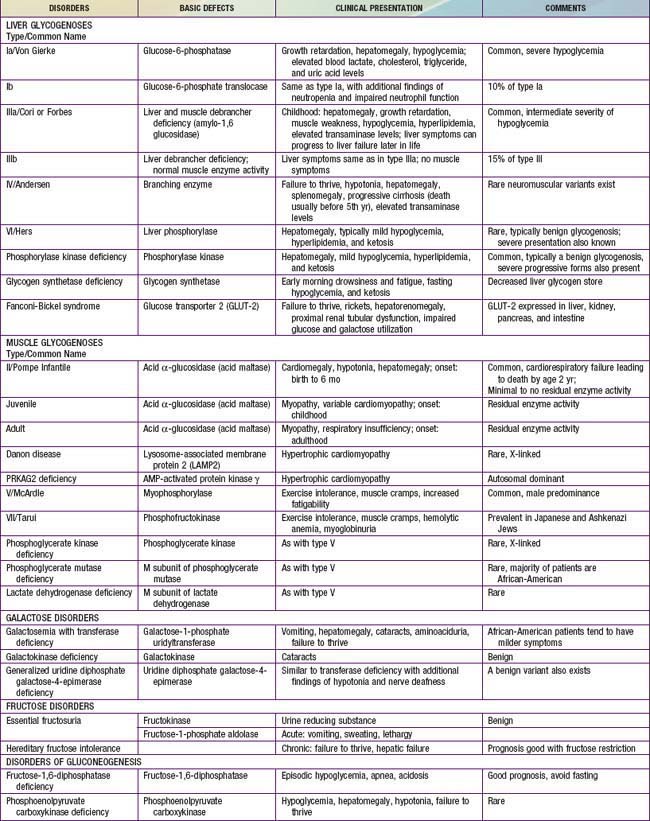
Defects in glycogen metabolism typically cause an accumulation of glycogen in the tissues, hence the name glycogen storage disease (Table 81-1). Defects in gluconeogenesis or the glycolytic pathway, including galactose and fructose metabolism, do not result in an accumulation of glycogen (see Table 81-1). The defects in pyruvate metabolism in the pathway of the conversion of pyruvate to carbon dioxide and water via mitochondrial oxidative phosphorylation are more often associated with lactic acidosis and some tissue glycogen accumulation.
81.1 Glycogen Storage Diseases
The disorders of glycogen metabolism, the glycogen storage diseases (GSDs), result from deficiencies of various enzymes or transport proteins in the pathways of glycogen metabolism (see Fig. 81-1). The glycogen found in these disorders is abnormal in quantity, quality, or both. GSDs are categorized by numeric type in accordance with the chronological order in which these enzymatic defects were identified. This numeric classification is still widely used, at least up to number VII. The GSDs can also be classified by organ involvement and clinical manifestations into liver and muscle glycogenoses (see Table 81-1).
Liver Glycogenoses
Type I Glycogen Storage Disease (Glucose-6-Phosphatase or Translocase Deficiency, Von Gierke Disease)
Treatment
Uncooked cornstarch acts as a slow-release form of glucose and can be introduced at a dose of 1.6 g/kg every 4 hr for infants <2 yr of age. The response of young infants is variable. As the child grows older, the cornstarch regimen can be changed to every 6 hr at a dose of 1.75-2.5 g/kg of body weight. New starch products, which are currently being developed, are thought to be longer acting, better tolerated, and more palatable. A short-term double-blind crossover pilot study comparing uncooked, physically modified cornstarch to traditional cornstarch showed that the majority of GSD patients treated with the new starch had better short-term metabolic control and longer duration of euglycemia; however, more extensive studies replicating these results are necessary at this time. Because fructose and galactose cannot be converted directly to glucose in GSD type I, these sugars are restricted in the diet. Sucrose (table sugar, cane sugar, other ingredients), fructose (fruit, juice, high fructose corn syrup), lactose (dairy foods), and sorbitol should be avoided or limited. Due to these dietary restrictions, vitamins and minerals such as calcium and vitamin D may be deficient and supplementation is required to prevent nutritional deficiencies. Dietary therapy improves hyperuricemia, hyperlipidemia, and renal function, slowing the development of renal failure. This therapy fails, however, to normalize blood uric acid and lipid levels completely in some individuals, despite good metabolic control, especially after puberty. The control of hyperuricemia can be further augmented by the use of allopurinol, a xanthine oxidase inhibitor. The hyperlipidemia can be reduced with lipid-lowering drugs such as HMG-CoA reductase inhibitors and fibrate (Chapter 80). Microalbuminuria, an early indicator of renal dysfunction in type I disease, is treated with angiotensin-converting enzyme (ACE) inhibitors. Citrate supplements can be beneficial for patients with hypocitraturia by preventing or ameliorating nephrocalcinosis and development of urinary calculi. Growth hormone should be used with extreme caution and limited to only those with a documented growth hormone deficiency. Even in those cases, there should be close monitoring of metabolic parameters and presence of adenomas.
Type III Glycogen Storage Disease (Debrancher Deficiency, Limit Dextrinosis)
Clinical Manifestations
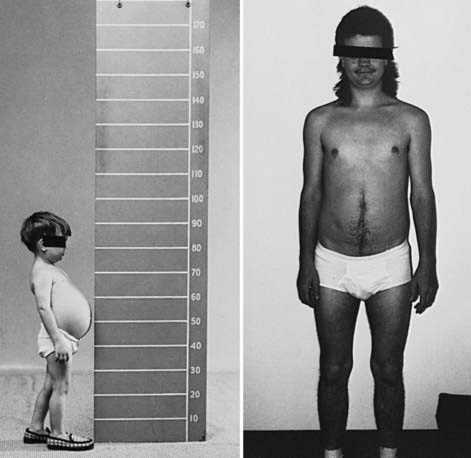
During infancy and childhood, the disease may be indistinguishable from type I GSD, because hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation are common (Fig. 81-2). Splenomegaly may be present, but the kidneys are not enlarged. Remarkably, hepatomegaly and hepatic symptoms in most patients with type III GSD improve with age and usually resolve after puberty. Progressive liver cirrhosis and failure can occur. Hepatocellular carcinoma has also been reported, more typically in patients with progressive liver cirrhosis. The frequency of adenomas in individuals with GSD III is far less, compared to GSD I. Furthermore, the relationship of hepatic adenomas and malignancy in GSD III is unclear. A single case of malignant transformation at the site of adenomas has been noted. In patients with muscular involvement (type IIIa), muscle weakness can present in childhood but can become severe after the 3rd or 4th decade of life, as evidenced by slowly progressive weakness and wasting. Because patients with GSD III can have decreased bone density, they are at an increased risk of potential fracture. Myopathy does not follow any particular pattern of involvement; both proximal and distal muscles are involved. Electromyography reveals a widespread myopathy; nerve conduction studies are often abnormal. Ventricular hypertrophy is a frequent finding, but overt cardiac dysfunction is rare. There have been some reports of life-threatening arrhythmia and need for heart transplant in some GSD III patients. Hepatic symptoms in some patients may be so mild that the diagnosis is not made until adulthood, when the patients show symptoms and signs of neuromuscular disease. The initial diagnosis has been confused with Charcot-Marie-Tooth disease. Polycystic ovaries are common; some patients can develop hirsutism, irregular menstrual cycles, and other features of polycystic ovarian syndrome. Fertility does not appear to be reduced; successful pregnancies in patients with GSD III have been reported.