Chapter 79 Defects in Metabolism of Amino Acids
79.1 Phenylalanine
Phenylalanine is an essential amino acid. Dietary phenylalanine not utilized for protein synthesis is normally degraded by way of the tyrosine pathway (Fig. 79-1). Deficiency of the enzyme phenylalanine hydroxylase (PAH) or of its cofactor tetrahydrobiopterin (BH4) causes accumulation of phenylalanine in body fluids and in the brain. The severity of hyperphenylalaninemia depends on the degree of enzyme deficiency and may vary from very high plasma concentrations (>20 mg/dL or >1,200 µmole/L, classic phenylketonuria [PKU]) to mildly elevated levels (2-6 mg/dL or 120-360 µmole/L). In affected infants with plasma concentrations >20 mg/dL, excess phenylalanine is metabolized to phenylketones (phenylpyruvate and phenylacetate; see Fig. 79-1) that are excreted in the urine, giving rise to the term phenylketonuria (PKU). These metabolites have no role in pathogenesis of central nervous system (CNS) damage in patients with PKU; their presence in the body fluids simply signifies the severity of the condition. The term hyperphenylalaninemia implies lower plasma levels (<20 mg/dL) of phenylalanine; these patients may or may not need dietary therapy based on their blood phenylalanine level. The brain is the main organ affected by hyperphenylalaninemia. The CNS damage in affected patients is caused by the elevated concentration of phenylalanine in brain tissue. The high blood levels of phenylalanine in PKU saturate the transport system across the blood-brain barrier causing inhibition of the cerebral uptake of other large neutral amino acids such as tyrosine and tryptophan. The exact mechanism of damage caused by elevated levels of intracerebral phenylalanine remains elusive. There are a few adults with classic PKU and normal intelligence who have never been treated with a phenylalanine-restricted diet. Phenylalanine content of the brain in these individuals was found to be close to that of normal subjects when studied by magnetic resonance spectroscopy (MRS) and imaging (MRI) techniques.
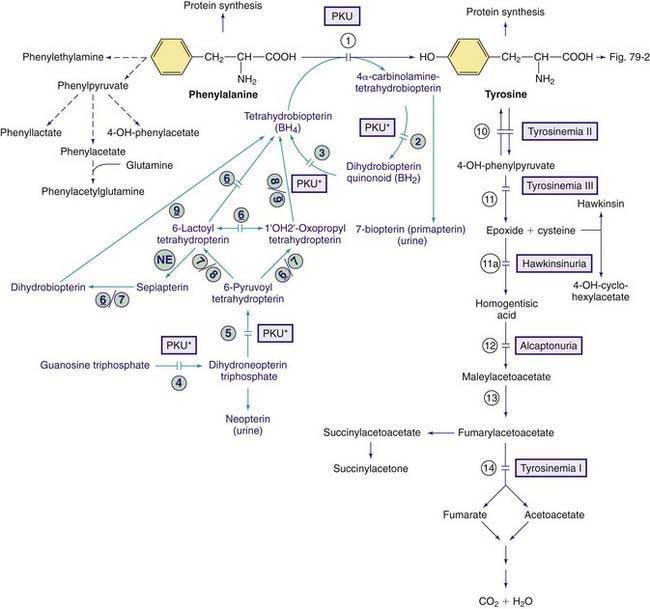
Figure 79-1 Pathways of phenylalanine and tyrosine metabolism. Enzyme defects causing genetic conditions are depicted as horizontal bars crossing the reaction arrow(s). Pathways for synthesis of cofactor BH4 are shown in purple. PKU* refers to defects of BH4 metabolism that affect the phenylalanine, tyrosine, and tryptophan hydroxylases (see Figs. 79-2 and 79-5). Enzymes: (1) phenylalanine hydroxylase (PAH), (2) pterin-carbinolamine dehydratase (PCD), (3) dihydrobiopterin reductase, (4) guanosine triphosphate (GTP) cyclohydrolase, (5) 6-pyruvoyltetrahydropterin synthase (6-PTS), (6) seriapterin reductase, (7) carbonyl reductase, (8) aldolase reductase, (9) dihydrofolate reductase, (10) tyrosine aminotransferase, (11a) intramolecular rearrangement, (11) 4-hydroxyphenylpyruvate dioxygenase, (12) homogentisic acid dioxygenase, (13) maleylacetoacetate isomerase, (14) fumarylacetoacetate hydroxylase, (NE) nonenzymatic.
Milder Forms of Hyperphenylalaninemia, Non-PKU Hyperphenylalaninemias
Hyperphenylalaninemia due to Deficiency of the Cofactor BH4
In 1-3% of infants with hyperphenylalaninemia, the defect resides in 1 of the enzymes necessary for production or recycling of the cofactor BH4 (Fig. 79-2). If these infants are misdiagnosed as having PKU, they may deteriorate neurologically despite adequate control of plasma phenylalanine. BH4 is synthesized from guanosine triphosphate (GTP) through several enzymatic reactions (see Fig. 79-1). In addition to acting as a cofactor for PAH, BH4 is also a cofactor for tyrosine hydroxylase and tryptophan hydroxylase, which are involved in the biosynthesis of dopamine (see Fig. 79-2) and serotonin (see Fig 79-5), respectively. Therefore, patients with hyperphenylalaninemia due to BH4 deficiency also manifest neurologic findings related to deficiencies of the neurotransmitters dopamine and serotonin. Four enzyme deficiencies leading to defective BH4 formation cause hyperphenylalaninemia and deficiencies of dopamine and serotonin. These include autosomal recessive GTP cyclohydrolase deficiency, pterin-carbinolamine dehydratase (PCD) deficiency, dihydropteridine reductase (DHPR) deficiency, and 6-pyruvoyltetrahydropterin synthase (PTPS or 6-PTS) deficiency. More than half of the reported patients have had a deficiency of 6-pyruvoyltetrahydropterin synthase. Autosomal dominant form of GTP deficiency and sepiapterin reductase deficiency result in deficiencies of neurotransmitters without hyperphenylalaninemia (Chapter 79.11 and Fig. 79-1).
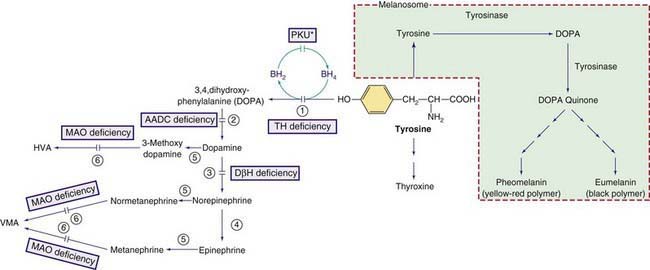
Figure 79-2 Other pathways involving tyrosine metabolism. PKU* indicates hyperphenylalanemia due to tetrahydrobiopterin (BH4) deficiency (see Fig. 79-1). HVA, homovanillic acid; VMA, vanillymandelic acid. Enzymes: (1) tyrosine hydroxylase (TH), (2) aromatic L-amino acid decarboxylase (AADC), (3) dopamine hydroxylase, (4) phenylethanolamine-N-methyltransferase (PNMT), (5) catechol O-methyltransferase (COMT), (6) Monoamine oxidase (MAO).
Diagnosis
BH4 deficiency and the responsible enzyme defect may be diagnosed by the following studies:
Blau N. Defining tetrahydrobiopterin (BH4)-responsiveness in PKU. J Inherit Metab Dis. 2008;31:2-3.
Blau N, Bélanger-Quintana A, Demirkol M, et al. Optimizing the use of sapropterin (BH4) in the management of phenylketonuria. Mol Genet Metab. 2009;96:158-163.
Blau N, Bélanger-Quintana A, Demirkol M, et al. Management of phenylketonuria in Europe: survey results from 19 countries. Mol Genet Metab. 2010;99:109-115.
Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417-1427.
Burton BK, Adams DJ, Grange DK, et al. Tetrahydrobiopterin therapy for phenylketonuria in infants and young children. J Pediatr. 2011;158:410-415.
Cederbaum S. Tetrahydrobiopterin and PKU: into the future. J Pediatr. 2011;158:351-353.
Committee on Genetics. Maternal phenylketonuria. Pediatrics. 2008;122:445-449.
Feillet F, van Spronsen FJ, MacDonald A, et al. Challenges and pitfalls in the management of phenylketonuria. Pediatrics. 2010;126:333-341.
Giovannini M, Verduci E, Salvatici E, et al. Phenylketonuria: dietary and therapeutic challenges. J Inherit Metab Dis. 2007;30:145-152.
Mitchell JJ, Scriver CR. Phenylalanine hydroxylase deficiency, 2010. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Scriver CR, Levy H, Donlon J. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2008.
Waisbren SE, Noel K, Fahrbach K, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol Genet Metab. 2007;92:63-70.
Waisbren SE, White D, van Spronsen F. Phenylketonuria, psychology and the brain. Mol Genet Metab. 2010;99(Suppl):S1-S108.
Zurfluh MR, Zschocke J, Lindner M, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008;29:167-175.
79.2 Tyrosine
Tyrosine is derived from ingested proteins or is synthesized endogenously from phenylalanine. It is used for protein synthesis and is a precursor of dopamine, norepinephrine, epinephrine, melanin, and thyroxine. Excess tyrosine is metabolized to carbon dioxide and water (see Fig. 79-1). Hereditary causes of hypertyrosinemia include deficiencies of tyrosine aminotransferase, 4-hydroxyphenylpyruvate dioxygenase (4-HPPD), or fumarylacetoacetate hydrolase (FAH). Acquired hypertyrosinemia may occur in severe hepatocellular dysfunction (liver failure), scurvy (vitamin C is the cofactor for the enzyme 4-HPPD), and hyperthyroidism. Hypertyrosinemia is common in blood samples obtained soon after eating.
Tyrosinemia Type I (Tyrosinosis, Hereditary Tyrosinemia, Hepatorenal Tyrosinemia)
Laboratory Findings
The presence of elevated levels of succinylacetone in serum and urine is diagnostic for tyrosinemia type I (see Fig. 79-1). In untreated patients, routinely available tests have a characteristic pattern. α-Fetoprotein level is increased, often markedly, and liver-synthesized coagulation factors are decreased in most patients; serum levels of transaminases are often increased, with marked increases being possible during acute hepatic episodes. Serum concentration of bilirubin is usually normal but can be increased with liver failure. Increased levels of α-fetoprotein are present in the cord blood of affected infants, indicating intrauterine liver damage. Plasma tyrosine level is usually elevated at diagnosis but this is a nonspecific finding and is dependent on dietary intake. Other amino acids, particularly methionine, may also be elevated in patients with liver damage. Hyperphosphaturia, hypophosphatemia, and generalized aminoaciduria may occur. The urinary level of 5-aminolevulinic acid is elevated (due to inhibition of 5-aminolevulinic hydratase by succinylacetone).
Diagnosis is usually established by demonstration of elevated levels of succinylacetone in urine or blood. Neonatal screening for hypertyrosinemia detects only a minority of patients with tyrosinemia type I. Succinylacetone, which is now assayed by most screening programs, has higher sensitivity and specificity than tyrosine and is the preferable metabolite for screening. Tyrosinemia type I should be differentiated from other causes of hepatitis and hepatic failure in infants, including galactosemia, hereditary fructose intolerance, neonatal iron storage disease, giant cell hepatitis, and citrullinemia type II (Chapter 79.11).
Treatment and Outcome
A diet low in phenylalanine and tyrosine can slow but does not halt the progression of the condition. The treatment of choice is nitisinone (NTBC), which inhibits tyrosine degradation at 4-HPPD (see Fig. 79-1). This treatment prevents acute hepatic and neurologic crises. Although nitisinone stops or greatly slows disease progression, some pretreatment liver damage is not reversible. Therefore, patients must be followed for development of cirrhosis or hepatocellular carcinoma. On imaging, the presence of even a single liver nodule usually indicates underlying cirrhosis. Most liver nodules in tyrosinemic patients are benign but current imaging techniques do not accurately distinguish all malignant nodules. Liver transplantation is an effective therapy for tyrosinemia type I and alleviates the risk of hepatocellular carcinoma. The impact of nitisinone treatment on the need for liver transplantation is still under study but the greatest effect is in patients treated early, such as children detected by neonatal screening, prior to the development of clinical symptoms. Rarely, nitisinone-treated patients develop corneal crystals, presumably of tyrosine, which are reversible by strict dietary compliance. This finding, combined with observations of developmental delay in some patients with chronically elevated tyrosine such as tyrosinemia type II, suggests that a diet low in phenylalanine and tyrosine should be continued in patients treated with nitisinone.
Tyrosinemia Type II (Richner-Hanhart Syndrome, Oculocutaneous Tyrosinemia)
This rare autosomal recessive disorder is caused by deficiency of tyrosine aminotransferase, resulting in palmar and plantar hyperkeratosis, herpetiform corneal ulcers, and mental retardation (see Fig. 79-1). Ocular manifestations include excessive tearing, redness, pain, and photophobia and often occur before skin lesions. Corneal lesions are presumed to be due to tyrosine deposition. In contrast to herpetic ulcers, corneal lesions in tyrosinemia type II stain poorly with fluorescein and often are bilateral. Skin lesions, which may develop later in life, include painful, nonpruritic hyperkeratotic plaques on the soles, palms and fingertips. Mental retardation, which occurs in <50% of patients, is usually mild to moderate.
The principal laboratory finding in untreated patients is marked hypertyrosinemia (20-50 mg/dL; 1,100-2,750 µmole/L). Surprisingly, 4-hydroxyphenylpyruvic acid and its metabolites are also elevated in urine despite being downstream from the metabolic block (see Fig. 79-1). This is hypothesized to occur via the action of other transaminases in the presence of high tyrosine concentrations, producing 4HPP in cellular compartments like the mitochondrion in which it is not further degraded. In contrast to tyrosinemia type I, liver and kidney function are normal, as are serum concentrations of other amino acids and succinylacetone. Tyrosinemia type II is due to TAT gene mutations, causing deficiency of cytosolic tyrosine aminotransferase activity in liver.
Transient Tyrosinemia of the Newborn
In a small number of newborn infants, plasma tyrosine may be as high as 60 mg/dL (3,300 µmole/L) during the 1st 2 weeks of life. Most affected infants are premature and are receiving high-protein diets. Transient tyrosinemia is felt to result from delayed maturation of 4-HPPD (see Fig. 79-1). Lethargy, poor feeding, and decreased motor activity are noted in some patients. Most are asymptomatic and are identified by a high blood phenylalanine or tyrosine level on screening. Laboratory findings include marked elevation of plasma tyrosine with a moderate increase in plasma phenylalanine. The finding of hypertyrosinemia differentiates this condition from PKU. 4-Hydroxyphenylpyruvic acid and its metabolites are present in the urine. Hypertyrosinemia usually resolves spontaneously in the 1st mo of life. It can be corrected promptly by reducing dietary protein to below 2 g/kg/24 hr and by administering vitamin C (200-400 mg/24 hr). Mild intellectual deficits have been reported in some infants that had this condition, but the causal relationship to hypertyrosinemia is not conclusively established.
Hawkinsinuria
This rare autosomal dominant condition is caused by a mutant 4-HPPD enzyme that catalyzes a partial reaction, releasing an intermediate compound used for diagnosis (see Fig. 79-1). This intermediate is either reduced to form 4-hydroxycyclohexylacetic acid (4-HCAA) or reacts with glutathione to form the unusual organic acid hawkinsin (2-L-cysteine-S-yl-1-4-dihydroxycyclohex-5-en-1-yl-acetic acid); secondary glutathione deficiency may occur.
Alcaptonuria
This rare (with an incidence of ≈1/250,000) autosomal recessive disorder is due to a deficiency of homogentisic acid oxidase, which causes large amounts of homogentisic acid to accumulate in the body and then to be excreted in the urine (see Fig. 79-1).
Albinism
Albinism is due to deficiency of melanin, the main pigment of the skin and eye (Table 79-1). Melanin is synthesized by melanocytes from tyrosine in a membrane-bound intracellular organelle, the melanosome. Melanocytes originate from the embryonic neural crest and migrate to the skin, eyes (choroid and iris), hair follicles, and inner ear. The melanin in the eye is confined to the retinal pigment epithelium, whereas in skin and hair follicles, it is secreted into the epidermis and hair shaft. Albinism can be caused by deficiencies of melanin synthesis, by some hereditary defects of melanosomes, or by disorders of melanocyte migration. Although albinism is a classical example of a biochemical genetic disease, neither the biosynthetic pathway of melanin nor many facets of melanocyte cell biology are completely elucidated (see Fig. 79-2). The end products are 2 pigments: pheomelanin, which is a yellow-red pigment; and eumelanin, a brown-black pigment.
Table 79-1 CLASSIFICATION OF ALBINISM
| TYPE | GENE | CHROMOSOME |
|---|---|---|
| OCULOCUTANEOUS ALBINISM (OCA) | ||
| OCA1 (tyrosinase deficient) | TYR | 11q |
| OCA1A (severe deficiency) | TYR | 11q |
| OCA1B (mild deficiency)* | TYR | 11q |
| OCA2 (tyrosinase positive)† | P (pink-eyed dilution) | 15q |
| OCA3 (Rufous, red OCA) | TYRP1‡ | 9p |
| OCA4 | MATP | 5p13.3 |
| Hermansky-Pudlak syndrome | HPS1 | 10q |
| Chédiak-Higashi syndrome | LYST | 1q |
| OCULAR ALBINISM | ||
| OA1 (Nettleship-Falls type) | OA | xp |
| LOCALIZED ALBINISM | ||
| Piebaldism | KIT | 4q |
| Waardenburg syndrome I & III | PAX3 | 2q |
| Waardenburg syndrome II | MITF | 3p |
* This includes Amish, minimal pigment, yellow albinism, and platinum and temperature-sensitive variants.
‡ Tyrosinase related protein 1.
Many clinical forms of albinism have been identified. Some of the seemingly distinct clinical forms are caused by different mutations of the same gene. Several genes located on different chromosomes are shown to be involved in melanogenesis (see Table 79-1). Attempts to differentiate types of albinism based on the mode of inheritance, tyrosinase activity, or the extent of hypopigmentation have failed to yield a comprehensive classification. The following classification is based on the distribution of albinism in the body and the type of mutated gene.
Mutation detection is clinically available for most albinism genes (see Table 79-1). Molecular diagnosis is of little use therapeutically in isolated albinism but can be helpful for precise genetic counseling of families.
Oculocutaneous (Generalized) Albinism (OCA)
OCA2 (Tyrosinase-Positive OCA)
This is the most common form of generalized OCA, particularly in African blacks. Clinically, patients demonstrate some pigmentation of the skin and eyes at birth and continue to accumulate pigment throughout their lives. The hair is yellow at birth and may darken with age. They have pigmented nevi and freckles but do not tan. They may be clinically indistinguishable from OCA1 B. These individuals have normal tyrosinase activity in hair bulbs. The defect is in the OCA2 gene on chromosome 15q, orthologous to the p (pink-eyed dilution) gene in the mouse. This gene produces the P protein, a melanosome membrane protein. Patients with Prader-Willi and Angelman syndromes with microdeletion in chromosome 15q12 lack 1 copy of the OCA2 gene and have mild pigmentary dilution (Chapter 76).
Syndromic Forms of Generalized Albinism
Chédiak-Higashi Syndrome
Patients with this rare autosomal recessive condition (Chapter 124) have albinism of variable severity and susceptibility to infection. Bacterial infections of skin and upper respiratory tract are common. Giant peroxidase-positive lysosomal granules can be seen in granulocytes in a blood smear. Patients have a reduced number of melanosomes, which are abnormally large (macromelanosomes). The bleeding tendency is typically mild. The major, life-threatening complication is macrophage activation with hemophagocytic lymphohistiocytosis, manifested by fever, lymphadenopathy, hepatosplenomegaly, cytopenias, and elevated plasma ferritin level. Patients surviving childhood may develop cerebellar atrophy, peripheral neuropathy, and cognitive delay. Mutations in the LYST gene on chromosome 1q are the only known cause of this syndrome.
Brilliant MH. Oculocutaneous albinism type 4, 2007. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
El-Karaksy H, Rashed M, El-Sayed R, et al. Clinical practice. NTBC therapy for tyrosinemia type 1: how much is enough? Eur J Pediatr. 2010;169:689-693.
Gahl WA. Hermansky-Pudlak syndrome, 2010. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Introne WJ, Westbroek W, Golas GA, et al. Chédiak-Higashi syndrome, 2009. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
King RA, Oetting W. Oculocutaneous albinism type 2, 2007. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis. 2008;31:81-87.
Pingault V, Ente D, Dastot-LeMoal F, et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391-406.
Rezvani I. 50 years ago in Journal of Pediatrics: Waardenburg’s syndrome. J Pediatr. 2010;157:732.
Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
79.3 Methionine
The normal pathway for catabolism of methionine, an essential amino acid, produces S-adenosylmethionine, which serves as a methyl group donor for methylation of a variety of compounds in the body, and cysteine, which is formed through a series of reactions collectively called trans-sulfuration (Fig. 79-3).
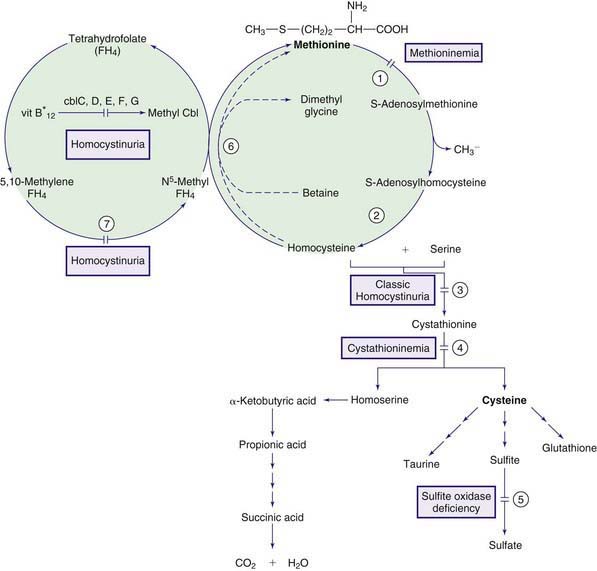
Homocystinuria (Homocystinemia)
Normally, most homocysteine, an intermediate compound of methionine degradation, is remethylated to methionine. This methionine-sparing reaction is catalyzed by the enzyme methionine synthase, which requires a metabolite of folic acid (5-methyltetrahydrofolate) as a methyl donor and a metabolite of vitamin B12 (methylcobalamin) as a cofactor (see Fig. 79-3). Only 20-30% of total homocysteine (and its dimer homocystine) is in free form in the plasma of normal individuals. The rest is bound to proteins as mixed disulfides. Three major forms of homocystinemia and homocystinuria have been identified.
Homocystinuria Due to Cystathionine β-Synthase (CBS) Deficiency (Classic Homocystinuria)
Infants with this disorder are normal at birth. Clinical manifestations during infancy are nonspecific and may include failure to thrive and developmental delay. The diagnosis is usually made after 3 yr of age, when subluxation of the ocular lens (ectopia lentis) occurs. This causes severe myopia and iridodonesis (quivering of the iris). Astigmatism, glaucoma, staphyloma, cataracts, retinal detachment, and optic atrophy may develop later in life. Progressive mental retardation is common. Normal intelligence has been reported. In an international survey of >600 patients, IQ scores ranged from 10 to 135. Higher IQ scores are seen in vitamin B6 responsive patients. Psychiatric and behavioral disorders have been observed in >50% of affected patients. Convulsions occur in about 20% of patients. Affected individuals with homocystinuria manifest skeletal abnormalities resembling those of Marfan syndrome (Chapter 693); they are usually tall and thin, with elongated limbs and arachnodactyly. Scoliosis, pectus excavatum or carinatum, genu valgum, pes cavus, high arched palate, and crowding of the teeth are commonly seen. These children usually have fair complexions, blue eyes, and a peculiar malar flush. Generalized osteoporosis, especially of the spine, is the main roentgenographic finding. Thromboembolic episodes involving both large and small vessels, especially those of the brain, are common and may occur at any age. Optic atrophy, paralysis, cor pulmonale, and severe hypertension (due to renal infarcts) are among the serious consequences of thromboembolism, which is caused by changes in the vascular walls and increased platelet adhesiveness secondary to elevated homocystine levels. The risk of thromboembolism increases after surgical procedures. Spontaneous pneumothorax and acute pancreatitis are rare complications.
Treatment with high doses of vitamin B6 (200-1,000 mg/24 hr) causes dramatic improvement in most patients who are responsive to this therapy. The degree of response to vitamin B6 treatment may be different in different families. Some patients may not respond because of folate depletion; a patient should not be considered unresponsive to vitamin B6 until folic acid (1-5 mg/24 hr) has been added to the treatment regimen. Restriction of methionine intake in conjunction with cysteine supplementation is recommended for patients who are unresponsive to vitamin B6. The need for dietary restriction and its extent remains controversial in patients with vitamin B6 responsive form. In some patients with this form, addition of betaine may obviate the need for any dietary restriction. Betaine (trimethylglycine, 6-9 g/24 hr for adults or 200-250 mg/kg/day for children) lowers homocysteine levels in body fluids by remethylating homocysteine to methionine (see Fig. 79-3); this may result in further elevation of plasma methionine levels. This treatment has produced clinical improvement (preventing vascular events) in patients who are unresponsive to vitamin B6 therapy. Cerebral edema has occurred in a patient with vitamin B6 nonresponsive homocystinuria and dietary noncompliance during betaine therapy. Administration of large doses of vitamin C (1 g/day) has improved the endothelial function; long-term clinical efficacy is not known.
Homocystinuria Due to Defects in Methylcobalamin Formation
Methylcobalamin is the cofactor for the enzyme methionine synthase, which catalyzes remethylation of homocysteine to methionine. There are at least 5 distinct defects in the intracellular metabolism of cobalamin that may interfere with the formation of methylcobalamin. To better understand the metabolism of cobalamin, see methylmalonic acidemia (Fig. 79-4; Chapter 79.6 and Fig. 79-3). The 5 defects are designated as cblC, cblD (including cblD variant 1), cblE (methionine synthase reductase), cblG (methionine synthase), and cblF. Patients with cblC, cblD (not those with cblD variant 2), and cblF defects have methylmalonic acidemia in addition to homocystinuria because formation of both adenosylcobalamin and methylcobalamin is impaired (Chapter 79.6).
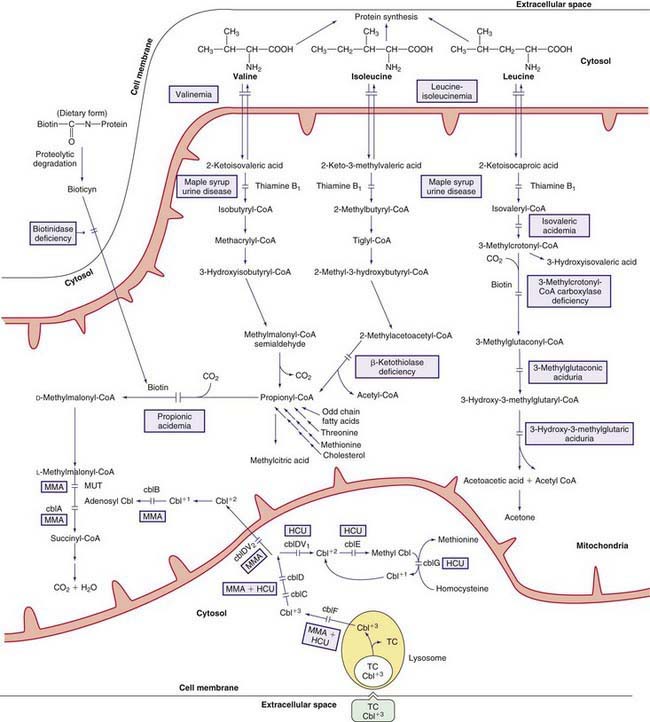
Patients with cblE and cblG defects are unable to form methylcobalamin and develop homocystinuria without methylmalonic acidemia (see Fig. 79-4); fewer than 40 patients are known with each of these diseases.
Hypermethioninemia
Secondary hypermethioninemia occurs in liver disease, tyrosinemia type I, and classic homocystinuria. Hypermethioninemia has also been found in premature and some full-term infants receiving high-protein diets, in whom it may represent delayed maturation of the enzyme methionine adenosyltransferase. Lowering the protein intake usually resolves the abnormality. Primary hypermethioninemia caused by the deficiency of hepatic methionine adenosyltransferase (MAT I/III; MAT II, which is present in other tissues, is not affected; see Fig. 79-3) has been reported in approximately 60 patients. The majority of these patients have been diagnosed in the neonatal period through screening for homocystinuria. Affected individuals with residual enzyme activity remain asymptomatic throughout life despite persistent hypermethioninemia. Some complain of unusual odor to their breath (boiled cabbage). A few patients with complete enzyme deficiency have had neurologic abnormalities related to demyelination (mental retardation, dystonia, dyspraxia). Normal pregnancies producing normal offspring have been reported in mothers with methionine adenoslytransferase deficiency. The condition is inherited as an autosomal recessive trait. The gene for hepatic methionine adenosyltransferase is on chromosome 10q22 and several disease-causing mutations have been identified. A novel defect, glycine N-methyltransferase deficiency, also causes isolated hypermethioninemia.
Adams D, Venditti CP. Disorders of intracellular cobalamin metabolism. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;30:827-834.
Forges T, Chery C, Audonnet S, et al. Life-threatening methylenetetrahydrofolate reductase (MTHFR) deficiency with extremely early onset: characterization of two novel mutations in compound heterozygous patients. Mol Genet Metab. 2010;100:143-148.
Holm PK, Hustad S, Ueland PM, et al. Modulation of the homocysteine-betaine relationship by methylenetetrahydrofolate reductase 677C-T genotypes and B-vitamin status in a large-scale epidemiologic study. J Clin Endocr Metab. 2007;92:1535-1541.
Lawson-Yuen A, Levy HL. The use of betaine in the treatment of elevated homocysteine. J Mol Genet Metab. 2006;88:201-207.
Lin HJ, Neidich JA, Salazar D, et al. Asymptomatic maternal combined homocystinuria and methylmalonic aciduria (cbIC) detected through low carnitine levels on newborn screening. J Pediatr. 2009;155:924-927.
Picker JD, Levy HL. Homocystinuria caused by cystathionine beta-synthase deficiency, 2006. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Schiff M, Benoist JF, Tilea B, et al. Isolated remethylation disorders: do our treatments benefit patients? J Inherit Metab Dis. 2011;34:137-145.
Skovby F, Gaustadnes M, Mudd SH. A revisit to the natural history of homocystinuria due to cystathionine beta-synthase deficiency. Mol Genet Metab. 2010;99:1-3.
Strauss KA, Morton DH, Puffenberger EG, et al. Prevention of brain disease from severe 5,10-methylenetetrahydrofolate reductase deficiency. Mol Genet Metab. 2007;91:165-175.
Testai FD, Gorelick PB. Inherited metabolic disorders and stroke part 2: homocystinuria, organic acidurias, and urea cycle disorders. Arch Neurol. 2010;67:148-153.
79.4 Cysteine/Cystine
Cysteine is a sulfur-containing nonessential amino acid that is synthesized from methionine (see Fig. 79-3). In the presence of oxygen, 2 molecules of cysteine are oxidized to form cystine. The most common disorders of cysteine/cystine metabolism, cystinuria (Chapter 541) and cystinosis (Chapter 523.3).
Sulfite Oxidase Deficiency (Molybdenum Cofactor Deficiency)
At the last step in cysteine metabolism, sulfite is oxidized to sulfate by sulfite oxidase, and the sulfate is excreted in the urine (see Fig. 79-3). This enzyme requires a molybdenum-pterin complex named molybdenum cofactor. This cofactor is also necessary for the function of 2 other enzymes in humans: xanthine dehydrogenase (which oxidizes xanthine and hypoxanthine to uric acid) and aldehyde oxidase. Three enzymes, encoded by 3 different genes, are involved in the synthesis of the cofactor. The genes are mapped to chromosomes 14q24, 6p21.3, and 5q11. Deficiency of any of the 3 enzymes causes cofactor deficiency with identical phenotype. Most patients who were originally diagnosed as having sulfite oxidase deficiency have been proven to have molybdenum cofactor deficiency. Both conditions are inherited as autosomal recessive traits. The gene for sulfite oxidase is on chromosome 12.
Kugler S, Hahnewald R, Garrido M, et al. Long-term rescue of a lethal inherited disease by adeno-associated virus-mediated gene transfer in a mouse model of molybdenum-cofactor deficiency. Am J Hum Genet. 2007;80:291-297.
Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460:839-847.
Tan WH, Eickler FS, Hoda S, et al. Isolated sulfite oxidase deficiency: a case report with a novel mutation and review of the literature. Pediatrics. 2005;116:757-766.
Veldman A, Santamaria-Araujo JA, Sollazzo S, et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics. 2010;125:e1249-e1254.
79.5 Tryptophan
Tryptophan is an essential amino acid and a precursor for nicotinic acid (niacin) and serotonin (Fig. 79-5). The genetic disorders of metabolism of serotonin, 1 of the major neurotransmitters, are discussed in Chapter 79.11.
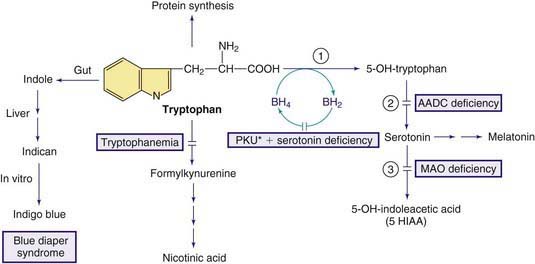
Figure 79-5 Pathways in the metabolism of tryptophan. PKU* indicates hyperphenylalanemia due to tetrahydrobiopterin deficiency (see Fig. 79-1). Enzymes: (1) tryptophan hydroxylase, (2) aromatic L-amino acid decarboxylase (AADC), (3) monoamine oxidase (MAO).
79.6 Valine, Leucine, Isoleucine, and Related Organic Acidemias
The early steps in the degradation of these 3 essential amino acids, the branched-chain amino acids, are similar (see Fig. 79-4). The intermediate metabolites are all organic acids, and deficiency of any of the degradative enzymes, except for the transaminases, causes acidosis; in such instances, the organic acids before the enzymatic block accumulate in body fluids and are excreted in the urine. These disorders commonly cause metabolic acidosis, which usually occurs in the 1st few days of life. Although most of the clinical findings are nonspecific, some manifestations may provide important clues to the nature of the enzyme deficiency. An approach to infants suspected of having an organic acidemia is presented in Figure 79-6. Definitive diagnosis is usually established by identifying and measuring specific organic acids in body fluids (blood, urine), by the enzyme assay, and by identification of the mutant gene.
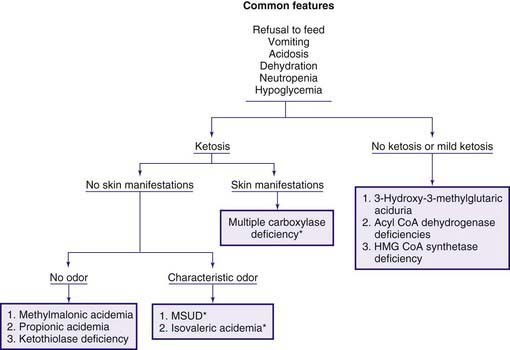
Figure 79-6 Clinical approach to infants with organic acidemia. Asterisks indicate disorders in which patients have a characteristic odor (see text and Table 79-2). MSUD, maple syrup urine disease.
Organic acidemias are not limited to defects in the catabolic pathways of branched-chain amino acids. Disorders causing accumulation of other organic acids include those derived from lysine (Chapter 79.14), those associated with lactic acid (Chapter 81), and dicarboxylic acidemias associated with defective fatty acid degradation (Chapter 80.1).
Maple Syrup Urine Disease (MSUD)
Decarboxylation of leucine, isoleucine, and valine is accomplished by a complex enzyme system (branched-chain α-ketoacid dehydrogenase) using thiamine pyrophosphate (vitamin B1) as a coenzyme. This mitochondrial enzyme consists of 4 subunits: E1α, E1β, E2, and E3. The E3 subunit is shared with 2 other dehydrogenases in the body, namely pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. Deficiency of this enzyme system causes MSUD (see Fig. 79-4), named after the sweet odor of maple syrup found in body fluids, especially urine. Based on clinical findings and response to thiamine administration, 5 phenotypes of MSUD have been identified.
Classic MSUD
Diagnosis is often suspected because of the peculiar odor of maple syrup found in urine, sweat, and cerumen (see Fig. 79-6). It is usually confirmed by amino acid analysis showing marked elevations in plasma levels of leucine, isoleucine, valine, and alloisoleucine (a stereoisomer of isoleucine not normally found in blood) and depression of alanine. Leucine levels are usually higher than those of the other 3 amino acids. Urine contains high levels of leucine, isoleucine, and valine and their respective ketoacids. These ketoacids may be detected qualitatively by adding a few drops of 2,4-dinitrophenylhydrazine reagent (0.1% in 0.1 N HCl) to the urine; a yellow precipitate of 2,4-dinitrophenylhydrazone is formed in a positive test. Neuroimaging during the acute state may show cerebral edema, which is most prominent in the cerebellum, dorsal brainstem, cerebral peduncle, and internal capsule. After recovery from the acute state and with advancing age, hypomyelination and cerebral atrophy may be seen in neuroimaging of the brain. The enzyme activity can be measured in leukocytes and cultured fibroblasts.



