Chapter 76 Cytogenetics
76.1 Methods of Chromosome Analysis
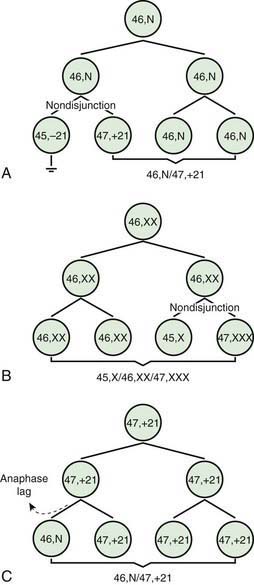
Two errors of cell division commonly occur during meiosis or mitosis, and either can result in an abnormal number of chromosomes. The 1st is nondisjunction, in which 2 chromosomes fail to separate during meiosis and thus migrate together into 1 of the new cells, producing 1 cell with 2 copies of the chromosome and another with no copy. The 2nd is anaphase lag, in which a chromatid or chromosome is lost during mitosis because it fails to move quickly enough during anaphase to become incorporated into 1 of the new daughter cells (Fig. 76-1).
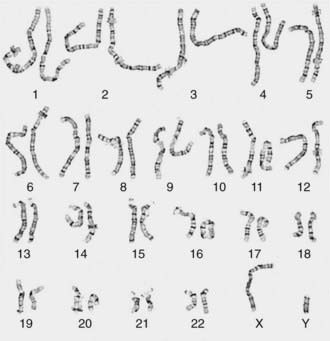
Other banding techniques such as Q-banding using quinacrine, reverse (R-banding) using acridine orange, and C-banding (constitutive heterchromatin) using barium hydroxide are available for use in certain circumstances but are losing ground to molecular technologies. Metaphase chromosome spreads are 1st evaluated microscopically, and then their images are photographed or captured by a video camera and stored on a computer to be later analyzed. Humans have 46 chromosomes or 23 pairs, which are classified as autosomes for chromosomes 1 to 22, and the sex chromosomes, often referred as sex complement: XX for females and XY for males. The homologous chromosomes from a metaphase spread can then be paired and arranged systematically to assemble a karyotype according to well-defined standard conventions like those established by International System for Human Cytogenetic Nomenclature (ISCN), with chromosome 1 being the largest and 22 the smallest. According to nomenclature, the description of the karyotype includes the total number of chromosomes followed by the sex chromosome constitution. A normal karyotype is 46,XX for females and 46,XY for males (Fig. 76-2). Abnormalities are noted after the sex chromosome complement.
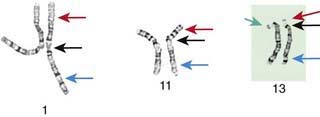
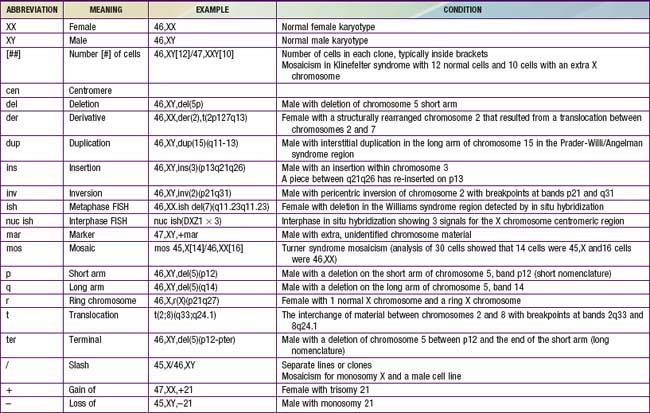
Although the internationally accepted system for human chromosome classification relies largely on the length and banding pattern of each chromosome, the position of the centromere relative to the ends of the chromosome also is a useful distinguishing feature (Fig. 76-3). The centromere divides the chromosome in 2, with the short arm designated as the p arm and the long arm designated as the q arm. A plus or minus sign before the number of a chromosome indicate that there is an extra or missing chromosome, respectively. Table 76-1 lists some of the abbreviations used for the descriptions of chromosomes and their abnormalities. A metaphase chromosome spread usually shows 450-550 bands. Prophase and prometaphase chromosomes are longer, are less condensed, and often show 550-850 bands. High-resolution analysis is useful for detecting subtle chromosome abnormalities that might otherwise go unrecognized.
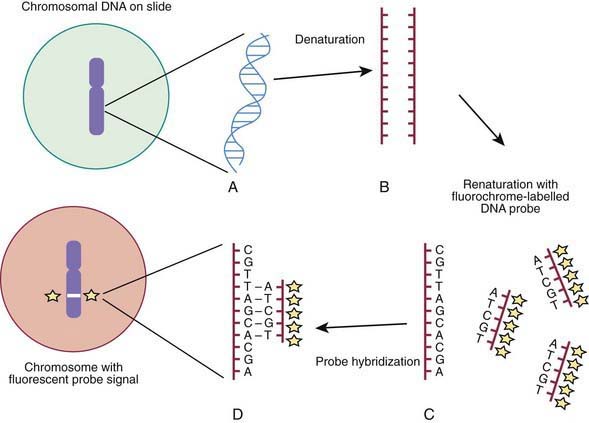
Molecular techniques such as fluorescence in situ hybridization (FISH) and comparative array genomic hybridization studies (conventional CGH and array CGH [aCGH]) have filled a significant void for the diagnosing cryptic chromosomal abnormalities. These techniques identify subtle abnormalities that are often below the resolution of standard cytogenetic studies. FISH is used to identify the presence, absence, or rearrangement of specific DNA segments and is performed with gene- or region-specific DNA probes. Several FISH probes are used in the clinical setting: unique sequence or single-copy probes, repetitive-sequence probes (alpha satellites in the pericentromeric regions), and multiple-copy probes (chromosome specific or painting). FISH involves using a unique known DNA sequence or probe labeled with a fluorescent dye that is complementary to the studied region of disease interest. The labeled probe is exposed to the DNA on a microscope slide, typically metaphase or interphase chromosomal DNA, that has been previously treated (denatured) to allow the DNA to become single stranded and to permit hybridization. When the probe pairs with its complementary DNA sequence, it can be then visualized by fluorescence microscopy (Fig. 76-4). In metaphase chromosome spreads, the exact chromosomal location of each probe copy can be documented and often the number of copies (deletions, duplications) of the DNA sequence as well, if they are not too close to each other; whereas in interphase cells, only the number of copies of a particular DNA segment can be determined. When the interrogated segments (as in genomic duplications) are close together, only interphase cells can accurately determine the presence of 2 or more copies or signals. In metaphase cells, some duplications might falsely appear as a single signal.
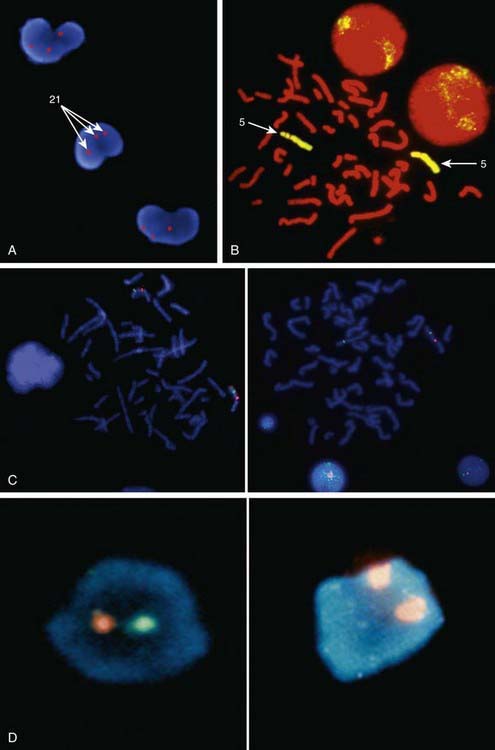
Metaphase and interphase FISH are particularly useful for detecting very small deletions that might escape notice with G-band analysis. In most cases the probe used for identification is used in conjunction with a control probe with a known location nearby the region studied. This allows correct identification of the hybridized signal to the right chromosome, and in some cases identification to the rearranged chromosome. With high-resolution chromosome analysis it is very difficult to recognize deletions of <5 million bp (5 Mb); FISH can reliably detect deletions as small as 50 to 200 kb of DNA. This has allowed the clinical characterization of a number of microdeletion syndromes. In addition to gene- or locus-specific probes, complex mixtures of DNA from a chromosome arm or an entire chromosome are available for fluorescence staining of large chromosome sections or entire chromosomes. The probe mixtures are referred to as chromosome paints (Fig. 76-5A and B). Other probes hybridize to repetitive sequences located to the pericentromeric regions. These probes are useful for the rapid identification of certain trisomies in interphase cells of blood smears, or even in the rapid analysis of prenatal samples from cells obtained through amniocentesis. Such probes are available for chromosomes 13, 18, and 21 and for the sex pair X and Y (Fig. 76-5C and D).
Comparative genomic hybridization (CGH) is a molecular-based technique that involves differentially labeling the patient’s DNA with a fluorescent dye (green) and a normal reference DNA with another fluorescent dye (red) (Fig. 76-6). Equal amounts of the two-label DNA samples are mixed and then used as a painting probe for FISH with normal metaphase chromosomes. The ratio of green : red fluorescence is measured along each chromosome. Regions of amplification of the patient’s DNA display an excess of green fluorescence, and regions of loss show excess red fluorescence. If the patient’s and the control DNA are equally represented, the green : red ratio is 1 : 1 and the chromosomes appear yellow.
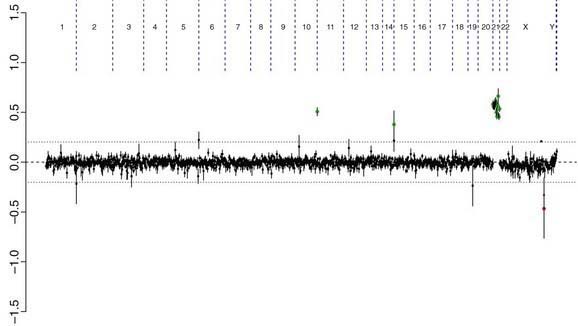
There are many copy number variations (CNVs) causing deletion or duplication in the human genome. Thus, most detected genetic abnormalities, unless associated with very well known clinical phenotypes, require parental investigations because a detected CNV that is inherited might turn out to be an incidental polymorphic variant. A de novo abnormality (i.e., 1 found only in the child and not the parents) is often more significant if it is associated with an abnormal phenotype and if it involves genes with important functions. aCGH is a very valuable technology alone or when combined with FISH and conventional chromosome studies (Fig. 76-7).
ACOG Practice Bulletin. Screening for fetal chromosomal abnormalities. Obstet Gynecol. 2007;109:217-227.
Bejjani BA, Saleki R, Ballif BC, et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet. 2005;134A:259-267.
Li MM, Andersson HC. Clinical application of microarray-based molecular cytogenetics: an emerging new era of genomic medicine. J Pediatr. 2009;155:311-317.
Saugier-Veber P, Girard-Lemaire F, Rudolf G, et al. Genetic compensation in a human genomic disorder. N Engl J Med. 2009;360:1211-1216.
76.2 Down Syndrome and Other Abnormalities of Chromosome Number
Aneuploidy and Polyploidy
The most common cause of aneuploidy is nondisjunction, the failure of chromosomes to disjoin normally during meiosis (see Fig. 76-1). Nondisjunction can occur during meiosis I or II or during mitosis. After meiotic nondisjunction, the resulting gamete either lacks a chromosome or has 2 copies instead of 1 normal copy, resulting in a monosomic or trisomic zygote, respectively.
FISH is a technique that can be used for rapid diagnosis in the prenatal detection of common fetal aneuploidies including chromosomes 13, 18, and 21 as well as sex chromosomes (see Fig. 76-5C and D). The most common numerical abnormalities in liveborn children include trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome), trisomy 13 (Patau syndrome), and sex chromosomal aneuploidies: Turner syndrome (usually 45,X), Klinefelter syndrome (47,XXY), 47,XXX, and 47,XYY, By far the most common type of trisomy in liveborn infants is trisomy 21 (47,XX,+21 or 47,XY,+21). Trisomy 18 and trisomy 13 are relatively less common and are associated with a characteristic set of congenital anomalies and severe mental retardation (Table 76-2). The occurrence of trisomy 21 and other trisomies increases with advanced maternal age (≥35 yr). Owing to this increased risk, women who are ≥35 yr at the time of delivery should be offered genetic counseling and prenatal diagnosis (including serum screening, ultrasonography, and amniocentesis or chorionic villus sampling; Chapter 90).
Table 76-2 CHROMOSOMAL TRISOMIES AND THEIR CLINICAL FINDINGS
Down Syndrome
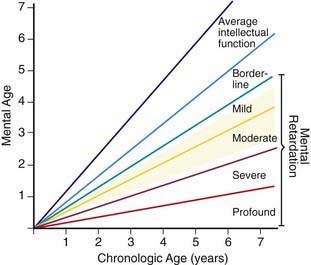
Trisomy 21 is the most common genetic cause of moderate mental retardation. The incidence of Down syndrome in live births is approximately 1 in 733; the incidence at conception is more than twice that rate; the difference is accounted by early pregnancy losses. In addition to cognitive impairment, Down syndrome is associated with congenital anomalies and characteristic dysmorphic features (Figs. 76-8 and 76-9; Table 76-3). Although there is variability in the clinical features, the constellation of phenotypic features is fairly consistent and permits clinical recognition of trisomy 21. Affected individuals are more prone to congenital heart defects (50%) such as atrioventricular septal defects, ventricular septal defects, isolated secundum atrial septal defects, patent ductus arteriosus, and tetralogy of Fallot. Congenital and acquired gastrointestinal anomalies and hypothyroidism are common (Table 76-4). Other abnormalities include megakaryoblastic leukemia, immune dysfunction, diabetes mellitus, and problems with hearing and vision (see Table 76-4). Alzheimer disease–like dementia is a known complication that occurs as early as the 4th decade and has an incidence 2-3 times higher than sporadic Alzheimer disease. Most males with Down syndrome are sterile, but some females have been able to reproduce, with a 50% chance of having trisomy 21 pregnancies. Two genes (DYRK1A, DSCR1) in the putative critical region of chromosome 21 may be targets for therapy.

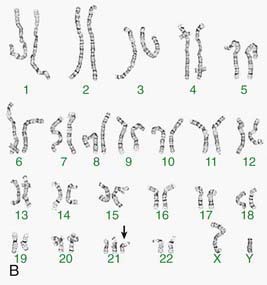
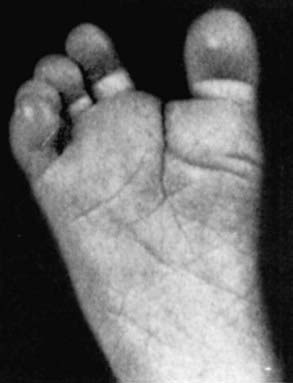
Figure 76-9 Prehensile foot in a 1-mo-old child.
(From Wiedemann HR, Kunze J, Dibbern H: Atlas of clinical syndromes: a visual guide to diagnosis, ed 3, St Louis, 1989, Mosby.)
Table 76-3 CLINICAL FEATURES OF DOWN SYNDROME IN THE NEONATAL PERIOD
CENTRAL NERVOUS SYSTEM
CRANIOFACIAL
CARDIOVASCULAR
MUSCULOSKELETAL
GASTROINTESTINAL
CUTANEOUS
Cutis marmorta
Table 76-4 ADDITIONAL FEATURES OF DOWN SYNDROME THAT CAN DEVELOP OR BECOME SYMPTOMATIC WITH TIME
NEUROPSYCHIATRIC
SENSORY
CARDIOVASCULAR
MUSCULOSKELETAL
ENDOCRINE
HEMATOLOGIC
GASTROINTESTINAL
CUTANEOUS
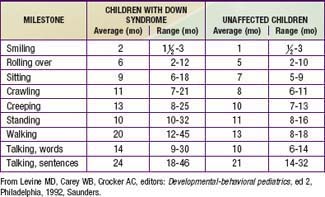
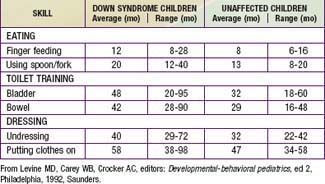
Developmental delay is universal (Tables 76-5 and 76-6; Fig. 76-10). Cognitive impairment does not uniformly affect all areas of development. Social development is relatively spared, but children with Down syndrome have considerable difficulty using expressive language. Understanding these individual developmental strengths will maximize the educational process for children with Down syndrome. Persons with Down syndrome often benefit from programs aimed at stimulation, development, and education. These programs are most effective in addressing social skills that often appear advanced for the intellectual delay. Children with Down syndrome also benefit from anticipatory guidance, which establishes the protocol for screening, evaluation, and care for patients with genetic syndromes and chronic disorders (Table 76-7).