Cyanotic Lesions
The differential diagnosis of cyanotic heart disease includes many disorders (Table 33-12). Lesions usually associated with decreased pulmonary flow include the tetralogy of Fallot, pulmonary stenosis, tricuspid atresia, pulmonary atresia with intact ventricular septum, and Ebstein disease. Cyanotic lesions usually associated with increased pulmonary vascular markings include d-transposition of the great arteries, hypoplastic left heart syndrome, total anomalous pulmonary veins, truncus arteriosus, and single ventricle.
Anomalies with Cyanosis Caused by Separate Transposed Systemic and Pulmonary Circulations
d-Transposition of the Great Arteries
With transposition of the great arteries, the aorta arises from the right ventricle and the pulmonary artery from the left ventricle. In the most common form, d-transposition, the aorta is anterior to and to the right of the pulmonary artery rather than in its normal rightward and posterior position.
Transposition of the great arteries is one of the most common congenital heart lesions presenting in the newborn period (see Tables 33-1 and 33-2), and is a frequent cause of death among un-operated neonates with congenital heart disease. The male-female ratio is 1.8:1, and the average birth weight is greater than that for other patients with congenital heart disease, although not for the general population. Transposition is associated with other cardiac abnormalities, including ventricular septal defect, patent ductus arteriosus, pulmonary valve stenosis, hypoplastic right ventricle, and coarctation.
Pathophysiology
The systemic and pulmonary circulations are normally in series with each other, but in complete transposition, the circulations are in parallel. Deoxygenated systemic venous blood returns to the right atrium, enters the right ventricle, and exits through the aorta. Maximally oxygenated pulmonary venous blood enters the left atrium and the left ventricle, and then returns to the pulmonary arteries and the lungs. Without some communication between the pulmonary and systemic circulations, survival is impossible; oxygenated blood cannot be delivered to the systemic circulation, nor can systemic venous blood be directed to the lung to become oxygenated. An atrial communication, ventricular defect, or patent ductus arteriosus, singly or in combination, may provide for mixing between the circulations (Fig. 33-16 and 33-17). The foramen ovale and ductus arteriosus, both normally patent in the fetus, usually close soon after birth. Infants with transposition and intact ventricular septum become extremely cyanotic within the first few hours or days after delivery, as closure of the foramen ovale and ductus arteriosus occurs and mixing between the circulations diminishes. The severe hypoxemia may lead to metabolic acidosis. Survival depends on prompt supportive medical care and reestablishment of patency of the ductus arteriosus and interatrial communication to improve mixing and oxygenation.
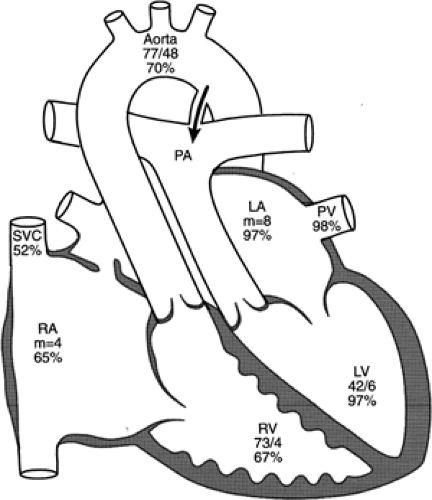 Figure 33-17 Diagram of the anatomy and physiology of transposition with intact ventricular septum in a 1-day-old girl who was cyanotic at birth. At catheterization, she had less than systemic pressure in the left (i.e., pulmonary) ventricle. The patent ductus arteriosus shunts blood into the pulmonary circuit, and the foramen ovale shunts an equal amount out of the pulmonary circuit. If this were not the case, relative blood volume would shift to one side of the circulation in a matter of minutes. If the ductus spontaneously closed, the infant’s condition would become precarious. If the ductus were dilated with prostaglandins, the infant would become pinker but might experience respiratory difficulty because of excess pulmonary flow. The numbers below the chamber name are pressure measurements (mm Hg) determined at cardiac catheterization; the percentages indicate oxygen saturation. LA, left atrium; LV, left ventricle; PA, pulmonary artery; PV, pulmonary vein; RA, right atrium; RV, right ventricle; SVC, superior vena cava. Adapted from Mullins CE, Mayer DC. Congenital heart disease: a diagramatic atlas. New York: Alan R Liss, 1988, with permission. |
Infants born with transposition and a large ventricular septal defect are less cyanotic because the ventricular defect allows mixing. These babies may not be recognized in the newborn period but appear in subsequent weeks with congestive failure. The combination of a large pulmonary flow, pulmonary hypertension, and elevation of left atrial pressure leads to the development of congestive heart failure (see Fig. 33-16) and later pulmonary vascular obstructive disease. Anatomic changes during the first few months of life may result in important hemodynamic changes. A large ventricular septal defect may spontaneously diminish in size or close, reducing mixing and increasing hypoxemia. Increasing pulmonary stenosis may decrease pulmonary flow and thereby increase cyanosis (See Color Plate) but improve congestive heart failure. Atrial septal defects created by balloon septostomy and those made by surgical septectomy may spontaneously diminish in size or close.
Clinical Findings
Infants with transposition of the great arteries and an intact ventricular septum develop marked cyanosis accompanied by mild tachypnea soon after birth. Often the infants, although tachypneic, do not seem distressed (i.e., peaceful cyanosis). The cardiac examination, chest radiograph, and ECG may otherwise be normal. The heart sounds are normal (i.e., the second heart sound splits), and there may be no significant murmur. Because the usual clinical findings, besides cyanosis, can be unremarkable, one of the most important diagnostic tests is the hyperoxia test. Failure of the arterial arterial oxygen pressure (PaO2) (often <30 mm Hg in room air) to rise significantly after the inhalation of 100% oxygen for a 10-minute period is strong presumptive evidence for cyanotic heart disease, most commonly complete transposition.
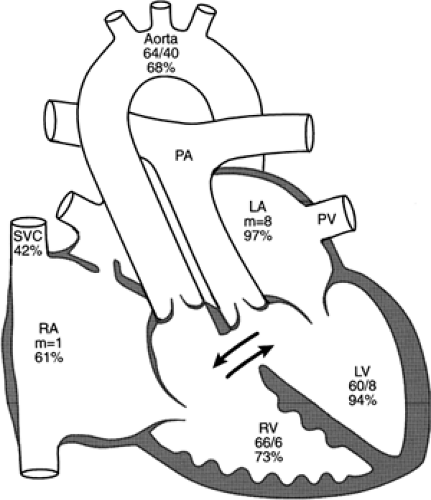 Figure 33-16 Diagram of the anatomy and physiology of transposition of the great arteries with a single membranous ventricular septal defect in a 1-month-old baby who had mild cyanosis and controlled congestive heart failure. There is equilibration of pressure between the ventricles and elevation of left-sided diastolic pressures. After balloon septostomy, the arterial saturation rose to 75%. The numbers below the chamber name are pressure measurements (mm Hg) determined at cardiac catheterization; the percentages indicate oxygen saturation. LA, left atrium; LV, left ventricle; PA, pulmonary artery; PV, pulmonary vein; RA, right atrium; RV, right ventricle; SVC, superior vena cava. Adapted from Mullins CE, Mayer DC. Congenital heart disease: a diagramatic atlas. New York: Alan R Liss, 1988, with permission. |
The ECG may show some excessive right ventricular forces. On chest radiograph the heart and pulmonary vascularity may initially appear normal, although cardiac enlargement, a narrow mediastinum, and pulmonary plethora are frequently present or develop.
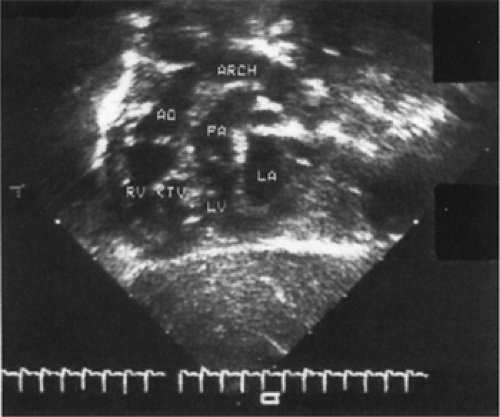 Figure 33-18 Echocardiographic subxiphoid view of a neonate with transposition of the great arteries. (AO aorta; ARCH, aortic arch; LA, left atrium; LV, left ventricle; PA, pulmonary artery; RV, right ventricle; TV, tricuspid valve.) |
Subcostal echocardiography reveals the diagnosis. The great artery arising from the left ventricle has an abnormal course and then bifurcates into the right and left pulmonary artery. The right ventricle gives rise to a great artery that passes relatively straight superiorly to the posterior arching aorta (Fig. 33-18). Echocardiographic examination can also determine the patency of the foramen ovale and ductus arteriosus, the nature of associated anomalies, and the coronary anatomy relevant to the surgical arterial switch procedure.
Infants with transposition and a large ventricular septal defect usually present with congestive failure and mild cyanosis, between zero and 6 weeks of age. Poor weight gain, tachypnea, and excessive diaphoresis are common, and wheezing occurs in older infants. A loud systolic murmur is present maximally at the lower left sternal border, often associated with a mid-diastolic flow rumble. An S3 may produce a gallop rhythm. Rales may be audible in the lungs.
ECG reveals right axis deviation and right atrial and right ventricular hypertrophy. Infrequently, if the right ventricle is hypoplastic, right ventricular forces may be absent or reduced, and left ventricular hypertrophy is present. Chest radiograph characteristically shows considerable cardiomegaly and pulmonary plethora.
Echocardiographic examination should identify the location of the ventricular septal defect, its relation to the great arteries and the atrioventricular valves, and complex associated problems including straddling or abnormal tricuspid valve, hypoplastic right ventricle, valvar or sub-valvar pulmonary stenosis, coarctation of the aorta, juxtaposition of the atrial appendages, and anomalous systemic or pulmonary venous drainage.
Differential Diagnosis
Most infants with transposition and intact ventricular septum are readily recognized as cyanotic infants with little respiratory distress and without significant murmur. Often a split second heart sound can be distinguished on exam, and chest radiograph demonstrates cardiomegaly and increased pulmonary flow, helping to distinguish transposition from other lesions with cyanosis and little murmur, pulmonary valve atresia with intact ventricular septum, and total anomalous pulmonary venous connection. Diagnosis of transposition of the great arteries can be complicated if other abnormalities, such as straddling tricuspid valve, hypoplastic right ventricle, coarctation of the aorta, or pulmonary stenosis, exist (see Table 33-12). Depending on the type and severity of the associated cardiac malformations, the clinical symptoms and findings in infants with complicated transposition of the great arteries may closely resemble those of almost any other cyanotic heart lesion.
The clinical picture in infants with transposition of the great arteries, ventricular septal defect, and pulmonary stenosis or atresia is virtually indistinguishable from that of tetralogy of Fallot or pulmonary atresia with a ventricular septal defect. If d-transposition of the great arteries is associated with a large ventricular septal defect and limited cyanosis, it is sometimes mistaken for other lesions with a large left-to-right shunt, such as a ventricular septal defect with normal aortic root or total anomalous pulmonary venous return without obstruction. The absence of cyanosis identifies the former, and echocardiography can differentiate all of these anomalies.
Treatment
In those with established or suspected transposition with intact ventricular septum, prostaglandin E1 is infused to open and maintain patency of the ductus arteriosus, to improve mixing and systemic oxygenation. Because prostaglandin E1 may cause apnea and vasodilation, support with mechanical ventilation, volume infusion, and sometimes inotropic agents may be required. Some babies also require a widely open atrial defect for adequate oxygenation, and most do better with one. Balloon atrial septostomy usually results in considerable clinical improvement in those sick from severe cyanosis, and some require it promptly for survival (see Fig. 33-19). The anatomy of the coronary arteries and associated lesions may also be established by catheterization. The type of surgical procedure employed depends on the associated cardiac defects. “Anatomic repair” with an arterial switch operation has been demonstrated to be the procedure of choice in neonates with uncomplicated transposition (56,57). The aorta and pulmonary arteries are transected, and the distal vessels are rejoined to provide normal physiologic connections. A button of proximal aortic tissue surrounding each coronary artery origin is cut, and both coronaries with the surrounding aortic button are moved from the native transposed aorta and attached to the neo-aorta. The ductus arteriosus is ligated, and the atrial septostomy is closed. Infants with transposition and isolated ventricular septal defect undergo arterial switch procedure and closure of the ventricular septal defect. Long-term outcome of arterial
switch procedure is not yet known but appears promising (58). Although a small number have difficulty with the coronary artery kinking, pulmonary artery compression, or anastomotic narrowing, the large majority do very well and lead essentially normal lives in childhood.
switch procedure is not yet known but appears promising (58). Although a small number have difficulty with the coronary artery kinking, pulmonary artery compression, or anastomotic narrowing, the large majority do very well and lead essentially normal lives in childhood.
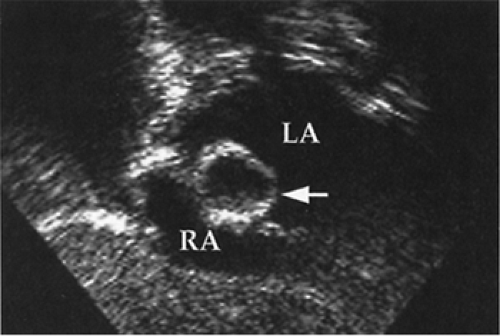 Figure 33-19 Echocardiographic subxiphoid view demonstrating a balloon septostomy catheter with the inflated balloon (arrow) being pulled from the left atrium (LA) to the right atrium (RA) through the foramen ovale. This septostomy technique is used in neonates with transposition of the great arteries to create an atrial septal defect to increase intracardiac mixing of fully and incompletely oxygenated blood, thereby improving systemic oxygenation. |
The additional presence of significant pulmonary or aortic valvar or sub-valvar stenosis can prohibit a straightforward arterial switch procedure. Severe pulmonary valve stenosis generally occurs with a large ventricular septal defect, and a Rastelli procedure or variation of it is done by placing a patch from the crest of the ventricular septum into the upper right ventricle to direct left ventricular blood to the transposed aorta and directing blood flow from the lower right ventricle to the pulmonary artery by interposition of a conduit (generally homograft) from a right ventriculotomy to the distal transected main pulmonary artery. The presence of native aortic valve stenosis can be dealt with by a modification of the Damus-Kaye-Stansel type in which left ventricular blood flow to the body is through anastomosis of the transected proximal main pulmonary artery to the ascending aorta, and pulmonary blood flow is through a conduit interposed between the right ventricle and distal main pulmonary artery. These procedures require revision of the surgically implanted conduits with growth and in the not infrequent occurrence of extrinsic compression. Without therapy, 95% of infants born with transpositions die within 1 year. With aggressive medical and surgical treatment, mortality is less than 5% to 10%.
Anomalies with Cyanosisfrom Decreased Pulmonary Blood Flow
Tetralogy of Fallot
The tetralogy of Fallot is characterized by a large ventricular septal defect and infundibular pulmonary stenosis or pulmonary atresia. Both environmental factors and a number of genetic disorders are associated with tetralogy of Fallot. Many have microdeletions in a critical region of chromosome 22q11 (25,30). The primary anatomic pathologic abnormality appears to be anterior deviation of the upper part of the ventricular septum, known as the conal septum that separates the anterior pulmonary outflow of the right ventricle from the sub-aortic left ventricular outflow. This results in the sub-aortic anterior malalignment ventricular septal defect and in the right ventricular infundibulum being hypoplastic and narrow. There is often considerable valvar pulmonary stenosis, hypoplasia of the pulmonary arteries, right ventricular hypertrophy, a relatively large ascending aorta, and a right aortic arch (25%). In infants with pulmonary atresia, pulmonary perfusion occurs by way of a patent ductus arteriosus or by systemic-to-pulmonary arterial collateral vessels. Five percent of patients have abnormal coronary distribution that may influence surgical correction. Tetralogy is one of the most common cyanotic congenital heart lesions presenting in the newborn period (see Tables 33-1 and 33-2) and is occasionally (10%) associated with severe extra-cardiac malformations.
Pathophysiology
Depending on the severity of right ventricular outflow obstruction, there may be intracardiac left-to-right flow or right-to-left shunt and hypoxemia. Pressures equalize between the ventricles through the large septal defect. The peripheral arterial oxygen saturation depends on the amount of systemic venous admixture and the absolute pulmonary flow (Fig. 33-20). The extent of systemic venous admixture, that is, right-to-left shunting of systemic venous blood away from the pulmonary outflow through the ventricular septal defect to the aorta, is directly related to the severity of the pulmonary stenosis and inversely related to the systemic vascular resistance. The amount of pulmonary blood flow depends on the amount of antegrade flow through the right ventricle outflow and the existence of alternative sources of flow (through a ductus arteriosus or systemic-to-pulmonary arterial collateral vessels). For example, in pulmonary atresia with a ventricular septal defect, the entire right heart output passes right to left through the ventricular defect, and pulmonary flow is supplied by a ductus arteriosus or collateral vessels and is usually less than normal. Cyanosis is the result. If the newborn has large aortic-pulmonary collateral vessels perfusing the lung, pulmonary blood flow may be large; and the infant may be barely cyanotic and some have congestive heart failure.
The pulmonary outflow stenosis in tetralogy of Fallot is progressive. Clinically significant cyanosis is present at birth in 25%, by 1 year of age in 75%, and almost all have become cyanotic by 20 years of age. Infundibular hypoplasia and stenosis are progressive in both absolute and relative terms. Progression of the stenosis during the first 6 months of life is most often largely relative because of a lack of adequate infundibular expansion during rapid somatic growth and need for proportionately greater pulmonary flow (59).
With time, complete atresia can occur. A patent ductus arteriosus usually closes within the first week of life, resulting in severe and often sudden hypoxemia or cyanotic spells.
With time, complete atresia can occur. A patent ductus arteriosus usually closes within the first week of life, resulting in severe and often sudden hypoxemia or cyanotic spells.
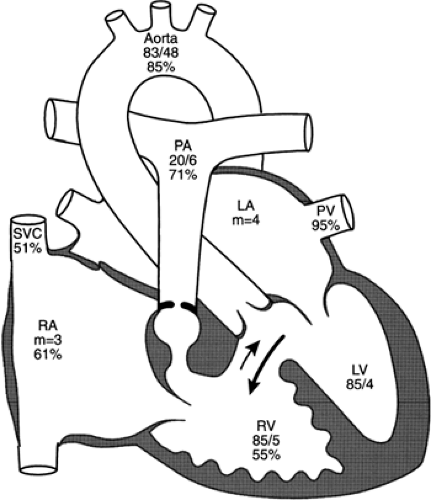 Figure 33-20 Diagram of the cardiac anatomy and physiology in a girl with mild cyanosis and a loud murmur audible at birth. She was found to have tetralogy of Fallot and was followed without medication prior to primary reparative surgery. The numbers below the chamber name are pressure measurements (mm Hg) determined at cardiac catheterization; the percentages indicate oxygen saturation. LA, left atrium; LV, left ventricle; PA, pulmonary artery; PV, pulmonary vein; RA, right atrium; RV, right ventricle; SVC, superior vena cava. (Adapted from Mullins CE, Mayer DC. Congenital heart disease: a diagramatic atlas. New York: Alan R Liss, 1988, with permission. |
Hypoxemia of rapid onset is characteristic of a “tet” spell secondary to an infundibular spasm. This may be associated with an increased adrenergic contractile state, systemic vasodilation associated with a meal, warm bath, or certain types of anesthesia, or constriction of a ductus arteriosus. Hypoxemia may result in a fall in systemic vascular resistance, metabolic acidosis, hyperpnea, and further hypoxemia. Hyperventilatory compensation for the metabolic acidosis may be ineffective because of inadequate pulmonary blood flow. The self-aggravating cycle of increasing hypoxemia and metabolic acidosis can progress to unconsciousness and convulsions.
Clinical Findings
Cyanosis of various degree and mild tachypnea often develop soon after delivery. If hypoxemia becomes severe, the infant may become hypotonic, hypotensive, and bradycardic. “Tet” spells characterized by a sudden onset of irritability, hyperpnea, and increasing cyanosis may develop. Spells may end in a loss of consciousness, seizures, cerebral injury, hemiparesis, or death. The disappearance of a previously heard right ventricular outflow systolic murmur with increased cyanosis suggests a spell and constitutes an indication for immediate therapy.
The second heart sound is single. With typical tetralogy, systolic murmurs from the pulmonary stenosis and/or ventricular septal defect are at the left upper and mid sternal border, respectively. With pulmonary atresia, when the systolic murmur is absent; there may be a constant apical systolic ejection click and prominent continuous murmurs of a patent ductus arteriosus or aortic-pulmonary collaterals, audible at the base, in the axillae, and/or over the back. A patent ductus arteriosus usually does not cause a continuous murmur in the first months of life; therefore, the presence of murmurs with cyanosis and a single S2 in a neonate strongly suggests tetralogy of Fallot with pulmonary atresia. Delay in height, weight, and skeletal maturation is common, particularly when DiGeorge syndrome is present, but some infants flourish despite severe hypoxemia.
Some young infants with the anatomic but acyanotic tetralogy of Fallot experience congestive heart failure from left-to-right ventricular septal shunting, later develop increased pulmonary stenosis, recover from congestion, and become cyanotic. Congestive heart failure also sometimes occurs in infants with pulmonary atresia when numerous or very large aortic-pulmonary collaterals are present. Although sub-acute bacterial endocarditis and brain abscess are common in older children with tetralogy of Fallot, these complications are extremely rare in infancy. However, spontaneous cerebrovascular accidents are common, particularly in infants with severe hypoxemia and relative anemia (<6-8 g/dL of oxyhemoglobin).
Chest radiograph shows a normal-sized heart, sometimes with right ventricular enlargement resulting in an upturned apex and an absent or diminished main pulmonary artery segment (i.e., boot-shaped heart), diminution of the pulmonary vasculature, and, in 25%, the aorta arching to the right.
ECG demonstrates right axis deviation, right atrial enlargement, and right ventricular hypertrophy, which at birth is often difficult to differentiate from normally prominent right ventricular forces.
Echocardiographic examination shows anterior and leftward deviation of the infundibular septum, creating sub-pulmonary stenosis and a malalignment ventricular septal defect with a large overriding aortic root (Fig. 33-21). Additional ventricular or atrial septal defects, central pulmonary artery hypoplasia, and coronary artery anatomy can usually be delineated by echocardiography but may sometimes require cardiac catheterization for clarification.
Angiography (Fig. 33-22), and often magnetic resonance imaging, may be used to determine presence and anatomy of distal pulmonary artery stenoses and systemic-arterial-to-pulmonary-arterial collaterals.
Differential Diagnosis
The features of cyanosis, a harsh systolic ejection murmur, chest radiographic findings of diminished pulmonary
vasculature with a normal-sized heart, and ECG evidence of right ventricular hypertrophy are characteristic of tetralogy of Fallot (see Table 33-12). The same findings with a continuous murmur suggest tetralogy of Fallot with pulmonary atresia. A few infants with tetralogy of Fallot and an underdeveloped pulmonary valve present with a characteristic to-and-fro murmur (i.e., steam engine sound) and severe respiratory distress caused by bronchial or tracheal compression by aneurysmally dilated pulmonary arteries.
vasculature with a normal-sized heart, and ECG evidence of right ventricular hypertrophy are characteristic of tetralogy of Fallot (see Table 33-12). The same findings with a continuous murmur suggest tetralogy of Fallot with pulmonary atresia. A few infants with tetralogy of Fallot and an underdeveloped pulmonary valve present with a characteristic to-and-fro murmur (i.e., steam engine sound) and severe respiratory distress caused by bronchial or tracheal compression by aneurysmally dilated pulmonary arteries.
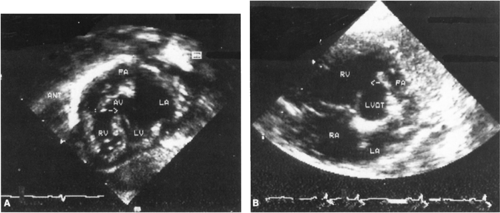 Figure 33-21 Echocardiographic subxiphoid (A) and parasternal short-axis (B) views of an infant with tetralogy of Fallot. Anterior deviation of the conal septum (above the arrow) is associated with malalignment ventricular septal defect and obstruction of the right ventricular outflow above the pulmonary artery; arrow, ventricular septal defect and narrowed right ventricular outflow; ANT, anterior; AV, aortic valve; LA, left atrium; LV, left ventricle; LVOT, left ventricular outflow tract; PA, pulmonary artery; RA, right atrium; RV, right venticle. |
Treatment
Initial treatment and timing of surgery depend on the severity of the pulmonary stenosis. If severe cyanosis develops in the newborn, prostaglandin E1 should be employed to reopen the ductus arteriosus, improve pulmonary perfusion and stabilize the infant, and surgery should be done. Cyanotic “tet” spells are much more likely to occur in the infant with a moderate or greater degree of preexisting cyanosis, and echocardiographic findings of severe sub-valvar infundibular stenosis and hypoplasia. Later onset cyanotic “tet” spells should be treated with oxygen, intramuscular or subcutaneous morphine sulfate (0.1 mg/kg), intravenous administration of saline boluses and sodium bicarbonate (approximately 1 mmol/kg), and, if needed, phenylephrine (0.1 mg/kg subcutaneously; 5 to 20 mg/kg by intravenous bolus; 0.1 to 0.5 mg/kg/min by intravenous infusion) titrated to elevate systemic vascular resistance and pressure. Propranolol may be of some value in treating the infant with a reactive infundibulum. The hemoglobin concentration should be maintained high enough to permit adequate oxygen transport. The occurrence of a single “tet” spell is an indication for surgery, possibly as an emergency procedure.
The newborn with tetralogy of Fallot and little or mild cyanosis can be carefully observed with repeated measurement of transcutaneous systemic oxygen saturation until a stable level is apparent after ductal closure. Many remain asymptomatic through the first 4 to 6 months of life, and can then undergo surgery with greater likelihood of preserving adequate pulmonary valve function. However, because the right ventricular outflow obstruction is often progressive, careful serial follow-up is prudent.
In the past, critically ill infants who required surgery underwent palliative procedures, usually a shunt between the subclavian artery and branch pulmonary artery (Blalock-Taussig). The shunt was ligated during a reparative operation when the child was older. Infants with uncomplicated tetralogy of Fallot requiring surgery now undergo one-stage reparative procedures with excellent results (60). Using sternotomy and cardiopulmonary bypass, the right ventricular outflow tract is enlarged with a pericardial patch, and the ventricular septal defect is closed with a Dacron patch. There are several potential late sequlae, including late dysrhythmias and problematic pulmonary regurgitation, but most patients with uncomplicated tetralogy of Fallot have an asymptomatic course into young-mid adulthood (61). Those with very low birth weight, anomalous origin of the left anterior descending coronary from the right coronary artery and those with pulmonary atresia and hypoplastic-distorted pulmonary arteries may require a palliative shunt with a more definitive repair, entailing a conduit from the right ventricle to the pulmonary artery, when the child is older.
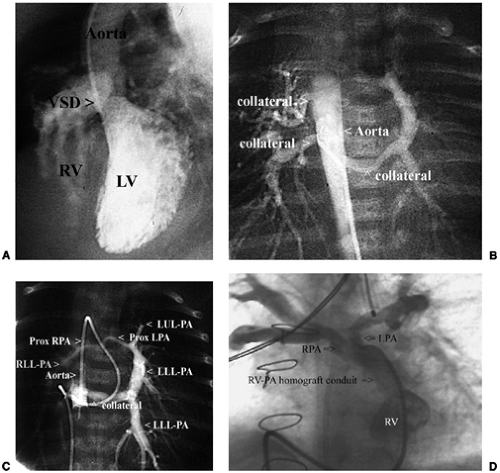 Figure 33-22 Angiograms from a cyanotic infant with complex tetralogy of Fallot with atresia of the right ventricular outflow, pulmonary valve and proximal main pulmonary artery demonstrate the anatomy of the hypoplastic mediastinal pulmonary arteries and collateral vessels that perfuse them. A. Left ventricular angiogram demonstrates tetralogy of Fallot with sub-aortic ventricular septal defect and pulmonary outflow atresia. B. Balloon occlusion angiography in the descending thoracic aorta demonstrates collateral vessels from the aorta to the right and left lung. C. Additional balloon occlusion aortography demonstates the collateral vessel to the left lower pulmonary artery is in continuity with hypoplastic central mediastinal and right pulmonary arteries. Left sublavian arteriography, not shown, demonstrated an additional collateral soley supplying much of the left upper lung lobe. D. Right ventricular angiography after surgical implantation of a homograft conduit from right ventricle to the central pulmonary artery, and catheter balloon angioplasties of the left and right pulmonary arteries demonstrates substantial increases in sizes of the pulmonary arteries. LV, left ventricle; LLL-PA, left lower lobe pulmonary artery; LPA, left pulmonary artery; LUL-PA, left upper lobe pulmonary artery; PA, central pulmonary artery; prox, proximal; RLL-PA, right lower lobe pulmonary artery; RPA, right pulmonary artery; RV, right ventricle; VSD, ventricular septal defect. |
In general, the earlier severe hypoxemia develops, the more severe is the tetralogy of Fallot, and the poorer the prognosis without surgery. The overall mortality rate without surgery was approximately 35% by 1 year of age.
Pulmonary Stenosis
Pulmonary Valve and Subvalvar Stenosis
Pulmonary valve stenosis is one of the more common intracardiac anomalies detected in the first month of life (Table 33-1). It is usually mild and not progressive. Even mild obstruction (<10 mm Hg systolic pressure gradient across the obstruction) produces a readily audible murmur. Moderate and severe pulmonary valve stenosis detected in the first week of life often progresses for a limited time over the following weeks or months. It generally presents with an isolated murmur radiating to the suprasternal notch, often but not always with an early systolic click resembling a split S1, little or no cyanosis, and no signs of congestive heart failure. A parasternal thrill, high-pitched systolic ejection murmur, and single second heart sound indicate severe pulmonary valve stenosis. Severe valvar obstruction may produce cyanosis (Fig. 33-23)
because of right-to-left shunting through a foramen ovale or rarely may present with the findings of right-sided congestive heart failure. In its most severe form, a very immobile, critically stenotic pulmonary valve requires ductal patency for adequate pulmonary blood flow and systemic arterial oxygenation. The right ventricular chamber may be small and noncompliant, resulting in some degree of right-to-left shunting although the foramen ovale, even after removal of the stenosis. Infundibular (i.e., sub-valvar) obstruction as an isolated lesion is rare; its presence usually indicates an associated ventricular defect.
because of right-to-left shunting through a foramen ovale or rarely may present with the findings of right-sided congestive heart failure. In its most severe form, a very immobile, critically stenotic pulmonary valve requires ductal patency for adequate pulmonary blood flow and systemic arterial oxygenation. The right ventricular chamber may be small and noncompliant, resulting in some degree of right-to-left shunting although the foramen ovale, even after removal of the stenosis. Infundibular (i.e., sub-valvar) obstruction as an isolated lesion is rare; its presence usually indicates an associated ventricular defect.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


