Organization of Critical Care
The concept and development of critical care for all aspects of medicine and surgery began in the 1960s. The National Institutes of Health held a Consensus Conference (1983) and the Society of Critical Care Medicine (1988, 1999) subsequently established guidelines for intensive care units (ICUs). Especially pertinent to obstetrics, costly ICUs prompted evolution of a step-down intermediate care unit. These units were designed for patients who did not require intensive care, but who needed a higher level of care than that provided on a general ward. The American College of Critical Care Medicine and the Society of Critical Care Medicine (1998) published guidelines for these units (Table 47-1).
TABLE 47-1. Guidelines for Conditions That Could Qualify for Intermediate Care
Cardiac: evaluation for possible infarction, stable infarction, stable arrhythmias, mild-to-moderate congestive heart failure, hypertensive urgency without end-organ damage
Pulmonary: stable patients for weaning and chronic ventilation, patients with potential for respiratory failure who are otherwise stable
Neurological: stable central nervous system, neuromuscular, or neurosurgical conditions that require close monitoring
Drug overdose: hemodynamically stable
Gastrointestinal: stable bleeding, liver failure with stable vital signs
Endocrine: diabetic ketoacidosis, thyrotoxicosis that requires frequent monitoring
Surgical: postoperative from major procedures or complications that require close monitoring
Miscellaneous: early sepsis, patients who require closely titrated intravenous fluids, pregnant women with severe preeclampsia or other medical problems
 Obstetrical Critical Care
Obstetrical Critical Care
Although the evolution of critical care for obstetrical patients has generally followed developments described above, there are no specific guidelines. Most hospitals employ a blend of these concepts, and in general, units can be divided into three types.
1. Medical or Surgical ICU—in most hospitals, severely ill women are transferred to a unit operated by medical and surgical “intensivists.” Admissions or transfers to these units are situation-specific and based on the acuity of care needed and on the ability of the facility to provide it. For example, in most institutions, pregnant women who require ventilatory support, invasive monitoring, or pharmacological support of circulation are transferred to an ICU. Another example is the neurological ICU (Sheth, 2012). A review of more than 25 tertiary-care referral institutions indicated that approximately 0.5 percent of obstetrical patients are transferred to these types of ICUs (Zeeman, 2006).
2. Obstetrical Intermediate Care Unit—sometimes referred to as a High-Dependency Care Unit (HDU)—an example of this system is the one at Parkland Hospital. Located within the labor and delivery unit, it has designated rooms and experienced personnel. The two-tiered system incorporates the guidelines for intermediate and intensive care. Care is provided by maternal-fetal medicine specialists and nurses with experience in critical care obstetrics. As needed, this team is expanded to include other obstetricians and anesthesiologists, gynecological oncologists, pulmonologists, cardiologists, surgeons, and other medical and surgical subspecialists. Many tertiary-care centers have developed similar intermediate care units and use selected triage to ICUs. Guidelines for such transfers must follow the federal Emergency Medical Treatment and Labor Act (EMTALA) guidelines. According to the American Academy of Pediatrics and the American College of Obstetricians and Gynecologists (2012), the minimal monitoring required for a critically ill patient during transport includes continuous pulse oximetry, electrocardiography, and regular assessment of vital signs. All critically ill patients must have secure venous access before transfer. Those who are mechanically ventilated must have their endotracheal tube position confirmed and secured before transfer. Left uterine displacement and supplemental oxygen should be applied routinely during transport of antepartum patients. The utility of continuous fetal heart rate or tocodynamic monitoring is unproven, therefore, its use should be individualized.
3. Obstetrical Intensive Care Unit—these units are full-care ICUs as described above, but are operated by obstetrical and anesthesia personnel in the labor and delivery unit. Very few units have these capabilities (Zeeman, 2003, 2006).
For smaller hospitals, transfer to a medical or surgical ICU may be preferable, and sometimes transfer to another hospital is necessary. As discussed, indications for admission to these various types of critical care units are highly variable. Shown in Table 47-2 are some examples. The American College of Obstetricians and Gynecologists (2011a) has summarized critical obstetrical care implementation depending on hospital size and technical facilities. Somewhat related, the concept has been explored of a medical emergency team—MET—for rapid response to emergent obstetrical situations (Gosman, 2008).
TABLE 47-2. Comparison of Acuity of Patient Mix for Obstetrical Critical Care Shown in Percent
 Pulmonary Artery Catheter
Pulmonary Artery Catheter
Data obtained during pregnancy with the pulmonary artery catheter (PAC) have contributed immensely to the understanding of normal pregnancy hemodynamics and pathophysiology of common obstetrical conditions. These include preeclampsia-eclampsia, acute respiratory distress syndrome (ARDS), and amnionic-fluid embolism (Clark, 1988, 1995, 1997; Cunningham, 1986, 1987; Hankins, 1984, 1985). Because of these studies, most have concluded that such monitoring is seldom necessary (American College of Obstetricians and Gynecologists, 2013; Dennis, 2012).
In nonobstetrical patients, randomized trials of nearly 5000 subjects have shown no benefits with pulmonary artery catheter monitoring (Harvey, 2005; Richard, 2003; Sandham, 2003). A randomized trial by the National Heart, Lung, and Blood Institute (2006b) assessed catheter-guided therapy in 1000 patients with ARDS. Invasive monitoring did not improve outcomes and had more complications. According to a recent Cochrane Database review, there have been no randomized trials using this for preeclampsia management (Li, 2012). Overall mechanisms, benefits, and risks were reviewed by Vincent (2011).
 Hemodynamic Changes in Pregnancy
Hemodynamic Changes in Pregnancy
Formulas for deriving some hemodynamic parameters are shown in Table 47-3. These measurements can be corrected for body size by dividing by body surface area (BSA) to obtain index values. Normal values for nonpregnant adults are used but with the caveat that these may not necessarily reflect changes induced by more “passive” uteroplacental perfusion (Van Hook, 1997).
TABLE 47-3. Formulas for Deriving Various Cardiopulmonary Parameters
Mean arterial pressure (MAP) (mm Hg) = [SBP + 2 (DBP)] ÷ 3
Cardiac output (CO) (L/min) = heart rate × stroke volume
Stroke volume (SV) (mL/beat) = CO/HR
Stroke index (SI) (mL/beat/m2) = stroke volume/BSA
Cardiac index (CI) (L/min/m2) = CO/BSA
Systemic vascular resistance (SVR) (dynes × sec × cm−5) = [(MAP − CVP)/CO] × 80
Pulmonary vascular resistance (PVR) (dynes × sec × cm−5) = [(MPAP − PCWP)/CO] × 80
In a landmark investigation, Clark and colleagues (1989) used pulmonary artery catheterization to obtain cardiovascular measurements in healthy pregnant women and again in these same women when nonpregnant. These values are shown in Chapter 4 (Table 4-4, p. 60) and are also plotted on a ventricular function curve (Fig. 4-9, p. 59). Because increased blood volume and cardiac output are compensated by decreased vascular resistance and increased pulse rate, ventricular performance remains within the normal range at term. A working knowledge of these changes is paramount to understanding the pathophysiology of pregnancy complications discussed later in this chapter and throughout the book.
ACUTE PULMONARY EDEMA
The incidence of pulmonary edema complicating pregnancy averages 1 in 500 to 1000 deliveries at tertiary referral centers. The two general causes are: (1) cardiogenic—hydrostatic edema caused by high pulmonary capillary hydraulic pressures and (2) noncardiogenic—permeability edema caused by capillary endothelial and alveolar epithelial damage. In pregnancy, noncardiogenic pulmonary edema is more common. Taken in toto, studies in pregnant women indicate that more than half who develop pulmonary edema have some degree of sepsis syndrome in conjunction with tocolysis, severe preeclampsia, or obstetrical hemorrhage combined with vigorous fluid therapy (Thornton, 2011). To categorize these differences, Dennis and Solnordal (2012) proposed a classification to include those women who are normotensive or hypotensive versus those with hypertension.
Although cardiogenic pulmonary edema is less frequent, common precipitating causes include resuscitation for hemorrhage and vigorous treatment of preterm labor. The causes in 51 women with pulmonary edema were cardiac failure, tocolytic therapy, iatrogenic fluid overload, or preeclampsia, approximately a fourth each (Sciscione, 2003). In another study, more than half of cases were associated with preeclampsia, and there was equal distribution of the other three causes (Hough, 2007). Although used less commonly today, tocolytic therapy with β-mimetic drugs at one time was the cause of up to 40 percent of pulmonary edema cases (DiFederico, 1998; Jenkins, 2003).
 Noncardiogenic Increased Permeability Edema
Noncardiogenic Increased Permeability Edema
Endothelial activation is the common denominator that is associated with preeclampsia, sepsis syndrome, and acute hemorrhage—or frequently combinations thereof—and they are the most common predisposing factors to pulmonary edema (Table 47-4). As discussed, these clinical scenarios are often associated with corticosteroids given to hasten fetal lung maturation along with vigorous fluid replacement and tocolytic therapy (Thornton, 2011). Although parenteral β-agonists are undisputedly linked to pulmonary edema, indictment of magnesium sulfate is less convincing. In one study in which pulmonary edema developed in 8 percent of nearly 800 women given magnesium sulfate for preterm labor, half of these were also given terbutaline (Samol, 2005). In our extensive experiences at Parkland Hospital with the use of magnesium sulfate for neuroprophylaxis for severe preeclampsia, we remain dubious that magnesium per se causes pulmonary edema. Similar conclusions were reached following a review by Martin and Foley (2006).
TABLE 47-4. Some Causes and Associated Factors for Pulmonary Edema in Pregnancy
Noncardiogenic permeability edema—endothelial activation with capillary-alveolar leakage:
Preeclampsia syndrome
Acute hemorrhage
Sepsis syndrome
Tocolytic therapy—β-mimetics, MgSO4
Aspiration pneumonitis
Vigorous intravenous fluid therapy
Cardiogenic pulmonary edema—myocardial failure with hydrostatic edema from excessive pulmonary capillary pressure:
Hypertensive cardiomyopathy
Obesity—adipositas cordis
Left-sided valvular disease
Vigorous intravenous fluid therapy
 Cardiogenic Hydrostatic Edema
Cardiogenic Hydrostatic Edema
Ventricular failure causing pulmonary edema in pregnancy is usually associated with some form of gestational hypertension. Although it can be due to congenital or acquired anatomical defects, diastolic dysfunction is frequently from chronic hypertension, obesity, or both (Jessup, 2003; Kenchaiah, 2002). In these women, acute systolic hypertension exacerbates diastolic dysfunction and causes pulmonary edema (Dennis, 2012; Gandhi, 2001). Of note, concentric and eccentric hypertrophy is two- to threefold more common in black women compared with white women (Drazner, 2005).
Despite an underlying cardiomyopathy, heart failure is commonly precipitated by preeclampsia, hypertension, hemorrhage and anemia, and puerperal sepsis (Cunningham, 1986; Sibai, 1987). In many of these, when echocardiography is done later, systolic function is normal as measured by ejection fraction, but evidence for diastolic dysfunction can often be found (Aurigemma, 2004). The use of brain natriuretic peptide (BNP) has not been evaluated extensively in pregnancy (Chap. 4, p. 61 and Appendix, p. 1291). This neurohormone is secreted from ventricle myocytes and fibroblasts with distention seen in heart failure. In nonpregnant patients, values < 100 pg/mL have an excellent negative-predictive value, and levels > 500 pg/mL have an excellent positive-predictive value. It is problematic that levels frequently are 100 to 500 pg/mL, and thus nondiagnostic (Ware, 2005). Values for N-terminal BNP and atrial natriuretic peptide (ANP) are both elevated with preeclampsia (Tihtonen, 2007).
 Management
Management
Acute pulmonary edema requires emergency management. Furosemide is given in 20- to 40-mg intravenous doses along with therapy to control dangerous hypertension. Further treatment depends on whether a woman is ante- or postpartum and whether the fetus is alive or not. A live fetus prohibits the use of cardioactive drugs that might lower peripheral resistance and in turn severely diminish uteroplacental circulation. The cause of cardiogenic failure is determined by echocardiography, which will help direct further therapy. Acute pulmonary edema is not, per se, an indication for emergency cesarean delivery. Indeed, in most cases, these women are better served by vaginal delivery.
ACUTE RESPIRATORY DISTRESS SYNDROME
Acute lung injury that causes a form of severe permeability pulmonary edema and respiratory failure is termed acute respiratory distress syndrome (ARDS). This is a pathophysiological continuum from mild pulmonary insufficiency to dependence on high inspired oxygen concentrations and mechanical ventilation. Because there are no uniform criteria for its diagnosis, the incidence is variably reported. One review computed it to be 1 in 3000 to 6000 deliveries, and this comports with our experiences at Parkland Hospital (Catanzarite, 2001). In its most extreme form requiring ventilatory support, there is an associated mortality rate of 45 percent. This rate can be as high as 90 percent if caused or complicated by sepsis (Phua, 2009). Although they are younger and usually healthier than the overall population, pregnant women still have mortality rates of 25 to 40 percent (Catanzarite, 2001; Cole, 2005). Finally, if ARDS develops antepartum, there is a correspondingly high perinatal mortality rate.
 Definitions
Definitions
Physiological criteria required for diagnosis of acute respiratory distress syndrome are study dependent. In general, less precise terms are used for clinical care. Most investigators have defined ARDS as radiographically documented pulmonary infiltrates, a ratio of arterial oxygen tension to the fraction of inspired oxygen (Pao2:Fio2) < 200, and no evidence of heart failure (Mallampalli, 2010). Recently, the international consensus-revised Berlin Definition was reported by the ARDS Definition Task Force (2012). It describes categories of mild (200 mm Hg < Pao2/Fio2 ≤ 300 mm Hg); moderate (100 mm Hg < Pao2/Fio2 ≤ 200 mm Hg); and severe (Pao2/Fio2 ≤ 100 mm Hg). To date, for most interventional studies, however, a working diagnosis of acute lung injury is made when the Pao2:Fio2 ratio is < 300 along with dyspnea, tachypnea, oxygen desaturation, and radiographic pulmonary infiltrates (Wheeler, 2007).
 Etiopathogenesis
Etiopathogenesis
ARDS is a pathophysiological description that begins with acute lung injury from various causes. Several disorders have been implicated as causes of acute pulmonary injury and permeability edema during pregnancy (Table 47-5). Although many are coincidental, some are more common in pregnant women. For example, in nonpregnant patients, sepsis and diffuse infectious pneumonia are the two most common single-agent causes and together account for 60 percent of cases. In pregnancy, however, pyelonephritis, chorioamnionitis, and puerperal pelvic infection are the most frequent causes of sepsis. As discussed previously, severe preeclampsia and obstetrical hemorrhage are also commonly associated with permeability edema. Importantly, more than half of pregnant women with acute respiratory distress syndrome have some combination of sepsis, shock, trauma, and fluid overload. With acute hemorrhage, the contribution of transfusion-related acute lung injury (TRALI) is unclear in obstetrical patients (Kopko, 2002). This entity is discussed further in Chapter 41 (p. 818).
TABLE 47-5. Some Causes of Acute Lung Injury and Respiratory Failure in Pregnant Women
Pneumonia—bacterial, viral, aspiration
Sepsis syndrome—chorioamnionitis, pyelonephritis, puerperal infection, septic abortion
Hemorrhage—shock, massive transfusion, transfusion-related acute lung injury (TRALI)
Preeclampsia syndrome
Tocolytic therapy
Embolism—amnionic fluid, trophoblastic disease, air, fat
Connective-tissue disease
Substance abuse
Irritant inhalation and burns
Pancreatitis
Drug overdose
Fetal surgery
Trauma
Sickle-cell disease
Miliary tuberculosis
Endothelial injury in the lung capillaries releases cytokines that recruit neutrophils to the site of inflammation. Here, they elaborate more cytokines to worsen tissue injury. There are three stages of ARDS development. First, the exudative phase follows widespread injury to microvascular endothelium, including the pulmonary vasculature, and there is also alveolar epithelial injury. These result in increased pulmonary capillary permeability, surfactant loss or inactivation, diminished lung volume, and vascular shunting with resultant arterial hypoxemia. Next, the fibroproliferative phase usually begins 3 to 4 days later and lasts up to day 21. Last, the fibrotic phase results from healing, and despite this, the long-term prognosis for pulmonary function is surprisingly good (Herridge, 2003; Levy, 2012).
 Clinical Course
Clinical Course
With pulmonary injury, the clinical condition depends largely on the insult magnitude, the ability to compensate for it, and the disease stage. For example, soon after the initial injury, there commonly are no physical findings except perhaps hyperventilation. And at first, arterial oxygenation usually is adequate. Pregnancy-induced mild metabolic alkalosis may be accentuated by hyperventilation. With worsening, clinical and radiological evidence for pulmonary edema, decreased lung compliance, and increased intrapulmonary blood shunting become apparent. Progressive alveolar and interstitial edema develop with extravasation of inflammatory cells and erythrocytes.
Ideally, pulmonary injury is identified at this early stage, and specific therapy is directed at the insult if possible. Further progression to acute respiratory failure is characterized by marked dyspnea, tachypnea, and hypoxemia. Additional lung volume loss results in worsening of pulmonary compliance and increased shunting. There are now diffuse abnormalities by auscultation, and a chest radiograph characteristically demonstrates bilateral lung involvement (Fig. 47-1). At this phase, the injury ordinarily would be lethal in the absence of high inspired-oxygen concentrations and positive airway pressure by mask or by intubation. When shunting exceeds 30 percent, severe refractory hypoxemia develops along with metabolic and respiratory acidosis that can result in myocardial irritability, dysfunction, and cardiac arrest.
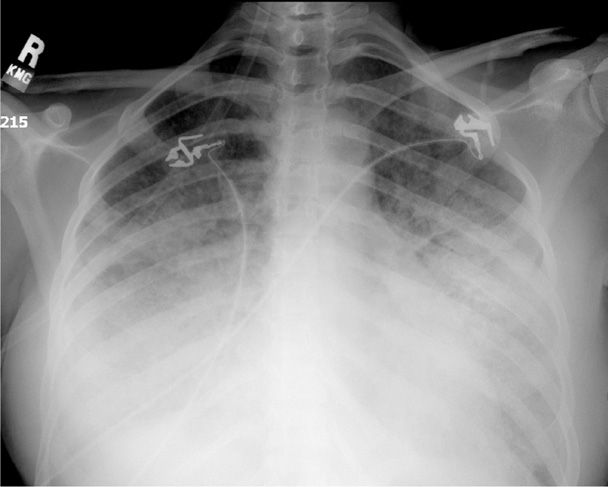
FIGURE 47-1 Anteroposterior chest radiograph of a second-trimester pregnant woman with marked bilateral parenchymal and pleural opacification secondary to acute respiratory distress syndrome (ARDS).
 Management
Management
In cases of severe acute lung injury, attempts are made to provide adequate oxygenation of peripheral tissues while ensuring that therapeutic maneuvers do not further aggravate lung injury. At least intuitively, increasing oxygen delivery should produce a corresponding increase in tissue uptake, but this is difficult to measure (Evans, 1999). Support of systemic perfusion with intravenous crystalloid and blood is imperative. As discussed on page 941, the trial conducted by the National Heart, Lung, and Blood Institute (2006b) showed that pulmonary artery catheterization did not improve outcomes. Because sepsis is commonplace in lung injury, vigorous antimicrobial therapy is given for infection, along with debridement of necrotic tissues. Oxygen delivery can be greatly improved by correction of anemia—each gram of hemoglobin carries 1.25 mL of oxygen when 90-percent saturated. By comparison, increasing the arterial Po2 from 100 to 200 mm Hg results in the transport of only 0.1 mL of additional oxygen for each 100 mL of blood.
Reasonable goals in caring for the woman with severe lung injury are to attain a Pao2 of 60 mm Hg or 90-percent saturation at an inspired oxygen content of < 50 percent and with positive end-expiratory pressures < 15 mm Hg. With regard to the pregnancy, it remains controversial whether delivery of the fetus improves maternal oxygenation (Cole, 2005; Mallampalli, 2010).
Mechanical Ventilation
Positive pressure ventilation by face mask may be effective in some women in early stages of pulmonary insufficiency (Roy, 2007). In an attempt to maximize the fetal environment, early intubation is preferred in the pregnant woman if respiratory failure is more likely than not, and especially if it appears imminent. There are many successful formulas for mechanical ventilation management (Levy, 2012). High-frequency oscillation ventilation (HFOV) has not been shown effective in ARDS (Ferguson, 2013; Slutsky, 2013; Young, 2013). Adjustments are made to obtain a Pao2 > 60 mm Hg or a hemoglobin saturation of ≥ 90 percent and a Paco2 of 35 to 45 mm Hg. Lower levels for Pao2 should be avoided, because placental perfusion may be impaired (Levinson, 1974).
For women who require ventilation for any length of time, the maternal mortality rate is approximately 20 percent. In a study of 51 such women, of whom almost half had severe preeclampsia, most required intubation postpartum. Eleven were delivered while being ventilated, and another six were discharged undelivered (Jenkins, 2003). There were two maternal deaths, including a woman who died as a complication of tocolytic treatment. In two other reports, maternal mortality rates were 17 and 25 percent (Chen, 2003; Schneider, 2003). None of these investigators concluded that delivery improved maternal outcome.
Positive End-Expiratory Pressure. With severe lung injury and high intrapulmonary shunt fractions, it may not be possible to provide adequate oxygenation with usual ventilatory pressures, even with 100-percent oxygen. Positive end-expiratory pressure is usually successful in decreasing the shunt by recruiting collapsed alveoli. At low levels of 5 to 15 mm Hg, positive pressure can typically be used safely. At higher levels, impaired right-sided venous return can result in decreased cardiac output, decreased uteroplacental perfusion, alveolar overdistention, falling compliance, and barotrauma (Slutsky, 2013).
Extracorporeal Membrane Oxygenation
As discussed in Chapter 33 (p. 638), extracorporeal membrane oxygenation (ECMO) has been successfully used for neonatal meconium aspiration syndrome. Preliminary observation suggests that it may be useful in adults (Brodie, 2011; Levy, 2012; Peek, 2009). ECMO has been used in pregnant women. In one study, 12 women with influenza-induced lung failure were treated with ECMO, and of four maternal deaths, three were due to anticoagulation-related hemorrhage (Nair, 2011). In other reports, ECMO was used to allow time for lung healing in five pregnant women, and the duration of support in the four survivors was 2 to 28 days (Cunningham, 2006). Technical aspects of ECMO were recently reviewed by Brodie and Bacchetta (2011).
Fetal Oxygenation
The propensity of the hemoglobin molecule to release oxygen is described by the oxyhemoglobin dissociation curve (Fig. 47-2). For clinical purposes, the curve can be divided into an upper oxygen association curve representing the alveolar-capillary environment and a lower oxygen dissociation portion representing the tissue-capillary environment. Shifts of the curve have their greatest impact at the steep portion because they affect oxygen delivery. A rightward shift is associated with decreased hemoglobin affinity for oxygen and hence increased tissue-capillary oxygen interchange. Rightward shifts are produced by hypercapnia, metabolic acidosis, fever, and increased 2,3-diphosphoglycerate levels. During pregnancy, the erythrocyte concentration of 2,3-diphosphoglycerate is increased by approximately 30 percent. This favors oxygen delivery to both the fetus and maternal peripheral tissues (Rorth, 1971).
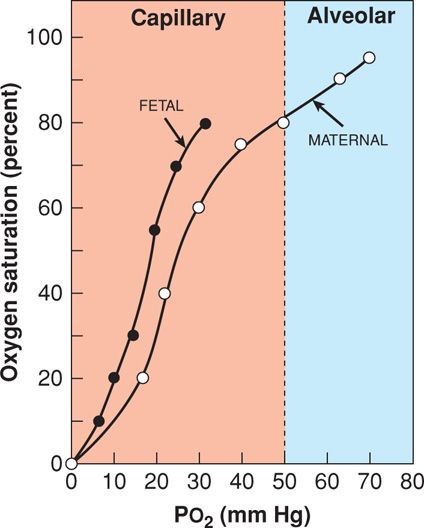
FIGURE 47-2 Oxyhemoglobin dissociation curve. With higher oxygen tension (Pao2) in the pulmonary alveoli, adult hemoglobin is maximally saturated compared with that at the lower oxygen tension in the tissue capillaries. Note that at any given oxygen tension, fetal hemoglobin carries more oxygen than adult hemoglobin, as indicated by percent saturation.
Fetal hemoglobin has a higher oxygen affinity than adult hemoglobin. As seen in Figure 47-2, its curve is positioned to the left of the adult curve. To achieve 50-percent hemoglobin saturation in the mother, the Pao2 must be 27 mm Hg compared with only 19 mm Hg in the fetus. Under normal physiological conditions, the fetus is constantly on the dissociation, or tissue, portion of the curve. Even with severe maternal lung disease and very low Pao2 levels, oxygen displacement to fetal tissues is favored. Another example of this comes from pregnant women who live at high altitudes, where despite a maternal Pao2 of only 60 mm Hg, the fetal Pao2 is equivalent to that of fetuses at sea level (Subrevilla, 1971).
Intravenous Fluids
Although mortality outcomes are similar, conservative versus liberal fluid management is associated with fewer days of mechanical ventilation (Wiedemann, 2006). There are some pregnancy-induced physiological changes that predispose to a greater risk of permeability edema from vigorous fluid therapy. Colloid oncotic pressure (COP) is determined by serum albumin concentration—1 g/dL exerts approximately 6 mm Hg pressure. As discussed in Chapter 4 (p. 67), serum albumin concentrations normally decrease in pregnancy. This results in a decline in oncotic pressure from 28 mm Hg in the nonpregnant woman to 23 mm Hg at term and to 17 mm Hg in the puerperium (Benedetti, 1979; Dennis, 2012). With preeclampsia, endothelial activation with leakage causes extravascular albumin loss and decreased serum albumin levels. As a result in these cases, oncotic pressure averages only 16 mm Hg antepartum and 14 mm Hg postpartum (Zinaman, 1985). These changes have a significant clinical impact on the colloid oncotic pressure/wedge pressure gradient. Normally, this gradient exceeds 8 mm Hg, however, when it is 4 mm Hg or less, there is an increased risk for pulmonary edema. These associations were recently reviewed by Dennis and Solnordal (2012).
 Other Therapy
Other Therapy
There were no benefits from artificial or replacement surfactant therapy in 725 nonpregnant patients with sepsis-induced lung failure (Anzueto, 1996). Although inhalation of nitric oxide was found to cause early improvement, mortality rates were unchanged in two studies (Taylor, 2004; Wheeler, 2007). In a trial conducted by the National Heart, Lung, and Blood Institute (2006a), prolonged methylprednisolone therapy did not reduce mortality rates.
 Long-Term Outcomes
Long-Term Outcomes
There are no long-term follow-up studies of pregnant women who recover from respiratory distress syndrome. In nonpregnant subjects, there are significant risks for impaired global cognitive function at 3 and 12 months (Pandharipande, 2013). Data from nonpregnant patients indicate a 1- to 2-year hiatus before basic normal activity is restored in all. In a 5-year follow-up study, Herridge and associates (2011) reported normal lung function but significant exercise limitation, physical and psychological sequelae, decreased physical quality of life, and increased costs and use of health-care services.
SEPSIS SYNDROME
Sepsis is derived from the ancient Greek sepein, “to rot.” The sepsis syndrome is induced by a systemic inflammatory response to bacteria or viruses or their by-products such as endotoxins or exotoxins. The severity of the syndrome is a continuum or spectrum (Fig. 47-3). Infections that most commonly cause the sepsis syndrome in obstetrics are pyelonephritis (Chap. 53, p. 1054), chorioamnionitis and puerperal sepsis (Chap. 37, p. 683), septic abortion (Chap. 18, p. 356), and necrotizing fasciitis (Chap. 37, p. 686). The mortality rate in nonpregnant patients is 20 to 35 percent with severe sepsis and 40 to 60 percent with septic shock (Angus, 2013; Munford, 2012). Mabie and coworkers (1997) reported a 28-percent mortality rate in 18 pregnant women with sepsis and shock. That said, the maternal mortality risk from sepsis is significantly underestimated (Acosta, 2013).
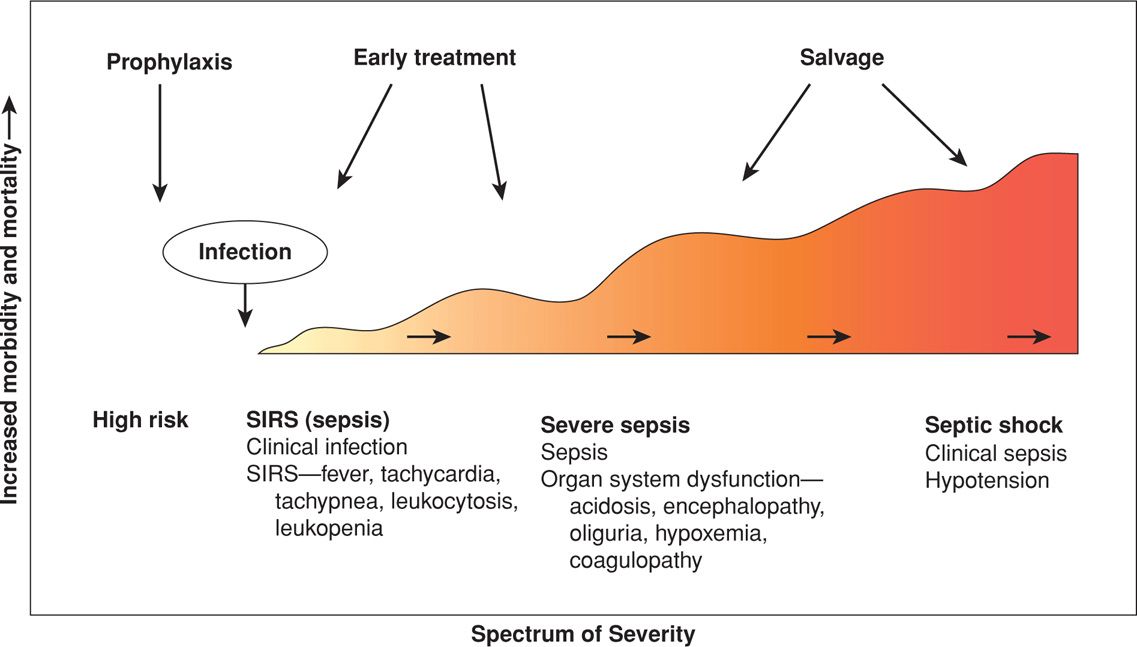
FIGURE 47-3 The sepsis syndrome begins with a systemic inflammatory response syndrome (SIRS) in response to infection that may progress to septic shock.
 Etiopathogenesis
Etiopathogenesis
Most of what is known concerning sepsis pathogenesis comes from study of lipopolysaccharide—LPS or endotoxin (Munford, 2012). The lipid A moiety is bound by mononuclear blood cells, becomes internalized, and stimulates release of mediators and a series of complex downstream perturbations. Clinical aspects of the sepsis syndrome are manifest when cytokines are released that have endocrine, paracrine, and autocrine effects (Angus, 2013).
Although the sepsis syndrome in obstetrics may be caused by several pathogens, most cases represent a small group. For example, pyelonephritis complicating pregnancy caused by Escherichia coli and Klebsiella species commonly is associated with bacteremia and sepsis syndrome (Cunningham, 1987; Mabie, 1997). And although pelvic infections are usually polymicrobial, bacteria that cause severe sepsis syndrome are frequently endotoxin-producing Enterobacteriaceae, most commonly E coli. Other pelvic pathogens are aerobic and anaerobic streptococci, Bacteroides species, and Clostridium species. Some strains of group A β-hemolytic streptococci and Staphylococcus aureus—including community-acquired methicillin-resistant strains (CA-MRSA)—produce a superantigen that activates T cells to rapidly cause all features of the sepsis syndrome—toxic shock syndrome (Moellering, 2011; Soper, 2011). This is discussed further in Chapter 37 (p. 690).
There are also potent bacterial exotoxins that can cause severe sepsis syndrome. Examples include exotoxins from Clostridium perfringens, toxic-shock-syndrome toxin-1 (TSST-1) from S aureus, and toxic shock-like exotoxin from group A β-hemolytic streptococci (Daif, 2009; Soper, 2011). These last exotoxins cause rapid and extensive tissue necrosis and gangrene, especially of the postpartum uterus, and may cause profound cardiovascular collapse and maternal death (Nathan, 1993; Sugiyama, 2010). In a review discussed subsequently, the maternal mortality rate from these infections was 58 percent (Yamada, 2010).
Thus, the sepsis syndrome begins with an inflammatory response that is directed against microbial endotoxins and exotoxins (Angus, 2013). CD4 T cells and leukocytes are stimulated to produce proinflammatory compounds that include tumor necrosis factor-α (TNF-α), several interleukins, other cytokines, proteases, oxidants, and bradykinin that result in a “cytokine storm” (Que, 2005; Russell, 2006). Many other cellular reactions then follow that include stimulation of pro- and antiinflammatory compounds, procoagulant activity, gene activation, receptor regulation, and immune suppression (Filbin, 2009; Moellering, 2011). It is also likely that IL-6 mediates myocardial suppression (Pathan, 2004).
The pathophysiological response to this cascade is selective vasodilation with maldistribution of blood flow. Leukocyte and platelet aggregation cause capillary plugging. Worsening endothelial injury causes profound permeability capillary leakage and interstitial fluid accumulation (Fig. 47-4). Depending on the degree of injury and inflammatory response, there is a pathophysiological and clinical continuum as depicted in Figure 47-3. The clinical syndrome begins with subtle signs of sepsis from infection and terminates with septic shock, which is defined by hypotension unresponsive to intravenous hydration. In its early stages, clinical shock results primarily from decreased systemic vascular resistance that is not compensated fully by increased cardiac output. Hypoperfusion results in lactic acidosis, decreased tissue oxygen extraction, and end-organ dysfunction that includes acute lung and kidney injury.
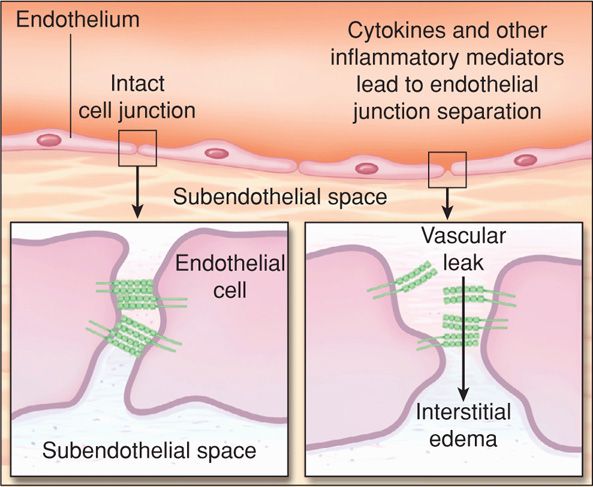
FIGURE 47-4 Endothelial permeability. The normal interendothelial interface is shown in the left inset. Cytokines and other inflammatory mediators disassemble the cellular junctions, resulting in microvascular leaks (right). (Modified from Lee, 2010.)
 Clinical Manifestations
Clinical Manifestations
The sepsis syndrome has myriad clinical manifestations that, at least in part, are dependent on the specific invading microorganism and its particular endo- or exotoxins. Some of the general effects of LPS are as follows:
1. Central nervous system: confusion, somnolence, coma, combativeness, fever, or hypoxemia
2. Cardiovascular: tachycardia, hypotension
3. Pulmonary: tachypnea, arteriovenous shunting with dysoxia and hypoxemia, exudative infiltrates from endothelial-alveolar damage, pulmonary hypertension
4. Gastrointestinal: gastroenteritis—nausea, vomiting, and diarrhea; hepatocellular necrosis—jaundice, hyperglycemia
5. Renal: prerenal oliguria, acute kidney injury
6. Hematological: leukocytosis or leukopenia, thrombocytopenia, activation of coagulation with disseminated intravascular coagulation
7. Cutaneous: acrocyanosis, erythroderma, bullae, digital gangrene.
Thus, although capillary leakage initially causes hypovolemia, if intravenous crystalloid is given at this point, then sepsis hemodynamically can be described as a high cardiac output, low systemic vascular resistance condition (Fig. 47-5). Concomitantly, pulmonary hypertension develops, and despite the high cardiac output, severe sepsis likely also causes myocardial depression (Ognibene, 1988). This is often referred to as the warm phase of septic shock. These findings are the most common cardiovascular manifestations of early sepsis, but they can be accompanied by some of the other clinical or laboratory aberrations listed above.
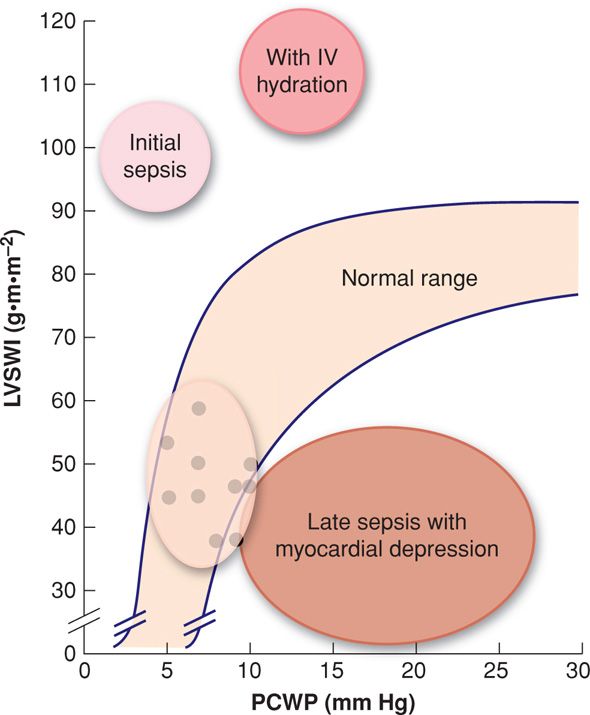
FIGURE 47-5 Hemodynamic effects of sepsis syndrome. Values for normal women at term are shown by dots. With early sepsis, there is high cardiac output and low vascular resistance. With fluid resuscitation, cardiac output increases even more, but so does capillary hydraulic pressure. With continued sepsis, there may be myocardial depression to further increase capillary hydraulic pressure. Decreased plasma oncotic pressure (serum albumin [g] × 6 mm Hg) contributes to interstitial lung fluid and endo/epithelial leak causes alveolar flooding. LVSWI = left ventricular stroke work index; PCWP = pulmonary capillary wedge pressure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


