Control of Breathing: Development, Apnea of Prematurity, Apparent Life-Threatening Events, Sudden Infant Death Syndrome
Mark W. Thompson
Carl E. Hunt
The fetus makes breathing movements in utero and these movements change in character and frequency throughout gestation (1). This “maturation” of breathing control is likely critical to initiation of spontaneous, regular respiratory effort at birth and normal control of postnatal breathing. Studies in animals and preterm infants have explored the complexities of this maturational process, but an exact timeline has yet to be delineated. In this chapter, normal control of breathing will first be reviewed, the “end-state” of this maturational process will be described, genetic influences on control of breathing will be summarized, and then what is known about fetal breathing, the effects of sleep on fetal breathing, and the factors involved with establishing normal patterns of breathing at birth will be reviewed. Finally, we will review apnea of prematurity (AOP), how this disorder may shed light on the normal maturational process of breathing control, how to treat AOP, and the potential adverse clinical consequences.
NORMAL CONTROL OF BREATHING
The control and maintenance of normal breathing largely reside within the respiratory control centers of the bulbopontine region of the brainstem. Neurons within this area of the brain respond to multiple afferent inputs to modulate their own inherent rhythmicity and provide efferent output to the respiratory control muscles. Multiple afferent inputs induce modulation of the central respiratory center efferent outputs to the respiratory and airway muscles and lungs. These afferent inputs are “categorized” by the respiratory control center; some inputs cause an instantaneous response in the control center output, while others only act to “shape” the respiratory response, resulting in small changes in muscular output, tidal volume, and airway tone (2). Among these inputs are signals from central and peripheral chemoreceptors, pulmonary stretch receptors, and cortical and reticuloactivating system neurons. These afferent inputs and resultant efferent outputs of the central respiratory center are summarized in Fig. 28-1. Sleep state can also have a profound effect on respiratory pattern.
In the adult, these multiple afferent inputs act upon the neurons within the respiratory control center and provide a well-integrated response to perturbations in the system and characteristic respiratory patterns. For example, increased respiratory rate and depth will characteristically result from activation of central chemoreceptors in response to hypercarbia. In the newborn infant and especially in the preterm infant, however, these responses are less well-organized and apnea is a common result of a disorganized response to multiple nonintegrated afferent inputs.
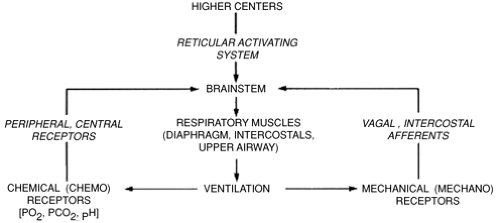 Figure 28-1 Major factors influencing respiratory control. PCO2, carbon dioxide partial pressure; PO2, oxygen pressure. (Reprinted from Martin RJ, Miller MJ, Carlo WA. Pathogenesis of apnea in preterm infants. J Pediatr 1986;109:733-741, with permission.) |
Genetic Influences
Sequencing the approximately 25,000 genes in the human genome has resulted in fundamental changes in our understanding of the role of specific gene products in the pathophysiology of all diseases. Partly related to studies of sudden infant death syndrome (SIDS) pathophysiology, recent genetic studies of cardiorespiratory regulation linking specific genotypes to specific components of brainstem autonomic regulation are also relevant to understanding control of breathing and its developmental regulation in general and to understanding the pathophysiology of AOP and apparent life-threatening events (ALTE) in particular (3).
Neural control of breathing and sleep are closely integrated, and abnormalities in regulation of sleep and circadian rhythmicity can impair cardiorespiratory integration and arousal responsiveness from sleep (4). Circadian rhythmicity has been extensively studied in animals, and homologous counterparts of essential circadian clock genes isolated in Drosophila have been identified in mammals (3). Since the sleep-wake cycle is under control of the circadian clock, these circadian master genes, as well as other sleep-related genes, likely influence sleep regulation.
Targeted gene inactivation studies in animals have identified several genes involved with prenatal brainstem development of respiratory control including arousal responsiveness (3). During embryogenesis, the survival of specific cellular populations composing the respiratory neuronal network is regulated by neurotrophins, a multigene family of growth factors and receptors. Brain-derived neurotrophic factor (BDNF) is required for development of normal breathing behavior in mice, and newborn mice lacking functional BDNF exhibit ventilatory depression associated with apparent loss of peripheral chemoafferent input (5). Ventilation is depressed and hypoxic ventilatory drive is deficient or absent.
Krox-20, a homeobox gene important for mouse hindbrain morphogenesis, also appears to be required for normal development of the respiratory central pattern generator (6). Krox-20-null mutants exhibit an abnormally slow respiratory rhythm and increased incidence of respiratory pauses, and this respiratory depression can be further modulated by endogenous enkephalins. Inactivation of Krox-20 may result in the absence of a rhythm-promoting reticular neuron group localized in the caudal pons and could thus be a cause of life-threatening apnea (3).
Brainstem muscarinic cholinergic pathways are important in ventilatory responsiveness to carbon dioxide (CO2) (3). The muscarinic system develops from the neural crest, and the ret proto-oncogene is important for this development. Ret knockout mice have a depressed ventilatory response to hypercarbia, implicating absence of the ret gene as a cause of impaired hypercarbic responsiveness. Diminished ventilatory responsiveness to hypercarbia has also been demonstrated in male newborn mice heterozygous for Mash-1 (7). There is a molecular link between ret and Mash-1, and the latter is expressed in embryonic neurons in vagal neural crest derivatives and in brainstem locus coeruleus neurons, an area involved with arousal responsiveness.
Serotonergic receptors in the brainstem are critical components of respiratory drive, and abnormalities have been implicated as causal in SIDS (8). Review of the genetics of serotonin metabolism may be very pertinent to understanding important genetic risk factors for SIDS, and serotonin metabolism may be a model for multiple other genetically derived neurotransmitter systems important in maturation and development of control of breathing. Serotonin is a widespread neurotransmitter affecting breathing, cardiovascular control, temperature, and mood (3,9). Serotonin modulates activity of the circadian clock and appears to be the major neurotransmitter of nonrapid eye movement (nonREM), or quiet, sleep. Many genes are involved in the control of serotonin synthesis, storage, membrane uptake, and metabolism. The serotonin transporter gene is located on chromosome 17, and variations in the promoter region of the gene appear to have a role in serotonin membrane uptake and regulation (3,9). Several transporter polymorphisms have been described, including one in the promoter region; the long “L” allele increases effectiveness of the promoter and hence leads to reduced serotonin concentrations at nerve endings compared to the short “S” allele. The L/L genotype is associated with increased serotonin transporters on neuroimaging, and increased postmortem binding. Although no studies have been done in AOP or ALTE subjects, recent data in white, African-American, and Japanese infants indicate that SIDS victims are more likely than controls to have the “L” allele (9,10).
Fetal Breathing
Fetal sheep make regular breathing movements during REM sleep (11). Studies in animal models and human fetuses have demonstrated that there is a maturational progression in fetal breathing movements over gestation and that this respiratory pattern can be altered by a variety of pharmacologic and physiologic inputs.
In the human fetus, ultrasound evidence of fetal breathing movements was first noted with development of high-speed ultrasound in the mid-1970s. These fetal breathing movements have been characterized throughout gestation
in response to maternal condition and as a potential indicator of overall fetal condition (12). From these studies, several characteristics of fetal breathing are evident. First, regardless of gestation, fetal breathing is not a continuous process. Significant periods of apnea, up to 122 minutes, are seen even in near-term fetuses (13). These periods of apnea are generally more frequent and of longer duration in younger gestational age fetuses (14), but significant respiratory pauses are still seen in presumed healthy near-term fetuses. Second, during periods of frequent respiratory motion, there are patterns of both regular and irregular breathing documented by chest/abdominal wall movements and by Doppler sonography assessing tracheal fluid flow (15). Third, there does appear to be a circadian rhythmicity to fetal breathing movements, with well-documented increases during certain periods of the day as compared to others (13). Fourth, maternal condition, particularly maternal glucose status, can have significant effects on fetal breathing frequency, with well-documented increases in fetal breathing frequency after maternal glucose loading following maternal fasting. This response to maternal glucose loading is not as pronounced when the mother is not fasting but rather ingesting normal meals during continuous 24-hour ultrasound monitoring (16). Fifth, fetal breathing does appear to be a potential measure of fetal well-being when used in conjunction with other ultrasound parameters. Several studies have documented diminished fetal breathing activity in association with poor fetal health, and this decreased activity, along with other measures of fetal well-being, can be helpful in guiding obstetric actions in terms of need for urgent delivery (17,18).
in response to maternal condition and as a potential indicator of overall fetal condition (12). From these studies, several characteristics of fetal breathing are evident. First, regardless of gestation, fetal breathing is not a continuous process. Significant periods of apnea, up to 122 minutes, are seen even in near-term fetuses (13). These periods of apnea are generally more frequent and of longer duration in younger gestational age fetuses (14), but significant respiratory pauses are still seen in presumed healthy near-term fetuses. Second, during periods of frequent respiratory motion, there are patterns of both regular and irregular breathing documented by chest/abdominal wall movements and by Doppler sonography assessing tracheal fluid flow (15). Third, there does appear to be a circadian rhythmicity to fetal breathing movements, with well-documented increases during certain periods of the day as compared to others (13). Fourth, maternal condition, particularly maternal glucose status, can have significant effects on fetal breathing frequency, with well-documented increases in fetal breathing frequency after maternal glucose loading following maternal fasting. This response to maternal glucose loading is not as pronounced when the mother is not fasting but rather ingesting normal meals during continuous 24-hour ultrasound monitoring (16). Fifth, fetal breathing does appear to be a potential measure of fetal well-being when used in conjunction with other ultrasound parameters. Several studies have documented diminished fetal breathing activity in association with poor fetal health, and this decreased activity, along with other measures of fetal well-being, can be helpful in guiding obstetric actions in terms of need for urgent delivery (17,18).
The fetal breathing pattern responds to a variety of pharmacologic and physiologic manipulations. Fetal breathing has been noted to increase three-fold if the mother inhales 5% CO2 for 15 minutes (19). Maternal hyperoxia does not alter fetal breathing movements or pattern in near-term normally grown fetuses (20), but growth-restricted fetuses do exhibit an increase in respiratory rate with maternal hyperoxia (21). Tocolytics, indomethacin, and terbutaline increase fetal breathing movements almost two-fold when administered to women in preterm labor between 26 and 32 weeks of gestation (22). Despite the increased sensitivity of ultrasound to detect and characterize fetal breathing movements, almost all of the studies described have been observational in nature and have lacked characterization of several other very important fetal physiologic correlates, such as diaphragmatic electrical activity and sleep state. These human ultrasound studies have also not addressed the effects of pharmacologic and physiologic manipulation on fetal breathing in a controlled fashion. Animal studies, however, do augment the human ultrasound data and provide a clearer picture of the maturational development of breathing control.
Fetal breathing activity in sheep starts early in gestation, arises from centrally mediated stimuli, and occurs primarily during periods of low-voltage electrocortical activity (REM sleep) (11). REM sleep comprises about 40% of fetal life during the last trimester in sheep. Breathing does occur during periods of high-voltage electrocortical activity (quiet sleep), but there is no significant pattern to this breathing—it is only episodic in nature and generally occurs after tonic muscular discharges, i.e., body movements (23).
Additional insights have been obtained using chronically instrumented fetal lambs with direct visualization of fetal breathing and state at baseline and after physiologic manipulations (23). Fetal sheep do not show any periods of wakefulness as indicated by eye opening and purposeful movement of the head. The fetus alternates between REM sleep (low-voltage electrocortical activity) and quiet sleep (high-voltage electrocortical activity). Breathing movements, swallowing, licking, and other body movements occur almost exclusively during REM sleep. The fetus responds to an increase in arterial carbon dioxide pressure (PaCO2) with an increase in breathing as determined by increases in tracheal pressure, breathing frequency, and integrated diaphragmatic activity, suggesting that both breathing rate and “tidal volume” increase in response to increasing hypercapnia (24,25,26). This breathing activity only occurs in REM sleep and during transitions to quiet sleep. Hypoxic levels of maternal oxygen abolish fetal breathing movements (27). These observations suggest that the relative low oxygen levels in the fetus at baseline contribute to the relative “lack” of fetal breathing movements throughout gestation and that the several-fold rise in oxygen partial pressure at birth is a contributing factor to the initiation of a regular respiratory pattern.
The use of pharmacologic agents in the fetal sheep model has also enhanced our understanding of fetal breathing. Indomethacin, pilocarpine, 5-hydroxytryptophan, and morphine all cause continuous breathing in both REM and quiet sleep. After these “breakthroughs” of breathing activity into quiet sleep, the intensity eventually diminished even when the fetus transitions back to REM sleep in association with decreasing drug levels (1,28).
In summary, animal experiments confirm that fetal breathing does occur. Under baseline conditions and nonpharmacologic manipulation (i.e., hypercarbia, hypoxia, hyperoxia), it occurs primarily during REM sleep (low-voltage electrocortical activity). The fetus does respond to central chemoreceptor input as evidenced by its response to hypercapnia. The fetal response to hypoxia appears to be centrally mediated and results in diminished normal neuronal activity and diminished or absent fetal breathing movements. The peripheral chemoreceptors may also be immature in the fetus and hence further contribute to absence of the “adult” response to hypoxia of an increase in respiratory rate and tidal volume.
Initiation of Breathing at Birth
Despite the enhanced understanding of fetal maturation of breathing control, the understanding remains incomplete regarding factors responsible for initiating and maintaining a regular pattern at birth. The traditional view is that mild fetal asphyxia during labor stimulates peripheral chemoreceptors leading to the first breath, and breathing is
then maintained via input of other sensory stimuli, e.g., cold and touch. However, this view has been challenged by animal experiments showing that denervation of the carotid and aortic chemoreceptors does not alter fetal breathing or the initiation/continuation of breathing at birth (29). A stable respiratory pattern can also be initiated in utero with cord occlusion and hyperoxia, independent of PaCO2. Postdelivery studies have also documented the importance of cooling in establishing a regular respiratory pattern and of CO2 in maintaining that pattern (30). There are some data to suggest that a placentally derived factor inhibits fetal breathing and removal of this factor at birth then permits initiation of a regular respiratory pattern (1). This placental inhibitor “theory” is intriguing, but it does not explain the totality of response needed to initiate and maintain a normal respiratory pattern at birth; also, no candidate inhibitor has been suggested. Development and maintenance of respiration in the newborn is more likely due to a complex interaction of sensory stimuli and both central and peripheral chemoreceptor inputs (Fig. 28-1). The degree of maturation in the central respiratory centers also appears to be important since the responses to these stimuli in the term infant are more developed than in the preterm infant.
then maintained via input of other sensory stimuli, e.g., cold and touch. However, this view has been challenged by animal experiments showing that denervation of the carotid and aortic chemoreceptors does not alter fetal breathing or the initiation/continuation of breathing at birth (29). A stable respiratory pattern can also be initiated in utero with cord occlusion and hyperoxia, independent of PaCO2. Postdelivery studies have also documented the importance of cooling in establishing a regular respiratory pattern and of CO2 in maintaining that pattern (30). There are some data to suggest that a placentally derived factor inhibits fetal breathing and removal of this factor at birth then permits initiation of a regular respiratory pattern (1). This placental inhibitor “theory” is intriguing, but it does not explain the totality of response needed to initiate and maintain a normal respiratory pattern at birth; also, no candidate inhibitor has been suggested. Development and maintenance of respiration in the newborn is more likely due to a complex interaction of sensory stimuli and both central and peripheral chemoreceptor inputs (Fig. 28-1). The degree of maturation in the central respiratory centers also appears to be important since the responses to these stimuli in the term infant are more developed than in the preterm infant.
The Preterm Infant
The respiratory responses to hypercarbia are diminished in preterm infants less than 33 weeks’ postconceptional age (PCA) (31), unrelated to any mechanical limitations in achieving adequate tidal volumes. The slope of the CO2 response curve (Fig. 28-2) increases significantly between 29 to 32 and 33 to 36 weeks of gestation. The cause of this decreased sensitivity of central respiratory centers to CO2 in preterm infants is related to central nervous system immaturity as indicated by decreased synaptic connections and incomplete dendritic arborization.
Preterm infants and full-term infants up to approximately 3 weeks postnatal age have a characteristic response to hypoxia that is quite different than in older infants. In contrast to the sustained hyperventilation in older infants, preterm infants have only a transient hyperventilation lasting 30 seconds to a minute. Subsequently, minute ventilation returns to baseline over several minutes and then progresses to ventilatory depression despite continued low inspired oxygen concentrations (1). The initial hyperventilatory response may be completely blunted in extremely premature infants (32). It is thought that this transient hyperventilation is in response to peripheral chemoreceptor input that is subsequently overridden by depression of central respiratory centers by hypoxia, and the hypoxic ventilatory depression is secondary to descending inhibition from the upper brainstem, midbrain, or higher structures (33,34).
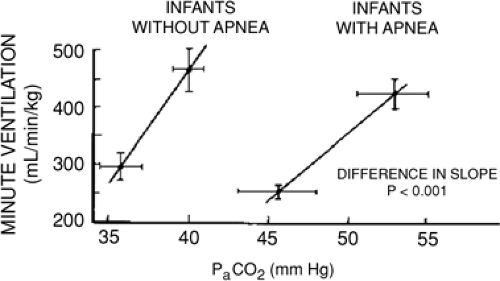 Figure 28-2 CO2 response curves for preterm infants with and without apnea of prematurity. PaCO2, arterial carbon dioxide pressure. (Reprinted from Gerhardt T, Bancalari E. Apnea of prematurity: I. Lung function and regulation of breathing. Pediatrics 1984; 74:58-62, with permission.) |
Upper airway reflexes also may play a role in inhibiting respiration, particularly in preterm infants. Multiple sensory afferent fibers exist within the upper airways, and stimulation of these fibers by various mechanisms can result in abnormal respiratory responses. Responses to upper airway afferent fiber stimulation can change markedly with maturation. Newborn rabbits, for example, can exhibit irreversible, fatal apnea with stimulation of the nares. This response does not occur in mature animals (34). Negative pressure in the upper airways in human infants results in depressed ventilation, and isolated application of negative pressure to the upper airway in a rabbit model can result in diaphragmatic inhibition (35). This inhibition may contribute to the centrally mediated apnea (central apnea) occurring in mixed apneas initiated by obstructed breaths. As upper airway obstruction occurs, the infant makes respiratory efforts against this obstruction and the resulting increased negative pressure in the upper airway may result in reflex inhibition of diaphragmatic contraction. Due to a blunted response to hypercarbia and hypoxic ventilatory depression, less mature preterm infants with apnea may be less able to recover spontaneously and hence more likely to require active intervention by health care providers.
Laryngeal reflexes can also have significant effects on control of breathing. Apnea has been associated with overt regurgitation of gastric contents into the upper airway (36). Either regurgitation or reflux of the acidic contents of the stomach into the area of the larynx appears to stimulate chemoreceptors in the receptor-rich area of the larynx. This leads to an inhibitory afferent signal to the central respiratory centers. Animal studies have demonstrated that the area of the larynx is rich in chemoreceptors connecting with afferent fibers leading back to the brainstem. Stimulation of these chemoreceptors results in apnea in a lamb model (37,38).
Periodic Breathing
Periodic breathing is defined as a pattern of breathing alternating with respiratory pauses lasting through three cycles of breathing, with the pauses or apneic periods lasting at least 3 seconds (1,39). Periodic breathing is frequently seen in preterm infants and also in term neonates and young infants.
Periodic breathing appears to occur predominantly during REM sleep, but it also occurs during quiet sleep (32,40). During quiet sleep, periodic breathing is “regular” with consistent durations of the apneic and breathing periods,
while during REM sleep periodic breathing tends to be irregular with inconsistent durations of cycles of periodic breathing. These episodes of periodic breathing are reflective of immaturity of the respiratory control system. While initially thought to be a “benign” respiratory pattern, increasing evidence has indicated that periodic breathing can have pathologic consequences. Particularly in more immature preterm infants, minute ventilation can diminish significantly during episodes of periodic breathing, and oxygen saturations can decrease to hypoxemic levels in association with longer apnea durations and increased time spent in periodic breathing. Since more immature infants generally spend more time in sleep and more of this sleep time is characterized by periodic breathing, these infants can spend a significant amount of time with low oxygen saturations. These extreme degrees of periodic breathing likely reflect a greater general immaturity of the central respiratory control centers; the more “severe” the periodic breathing, the more likely an infant is to demonstrate overt clinical symptoms related to AOP.
while during REM sleep periodic breathing tends to be irregular with inconsistent durations of cycles of periodic breathing. These episodes of periodic breathing are reflective of immaturity of the respiratory control system. While initially thought to be a “benign” respiratory pattern, increasing evidence has indicated that periodic breathing can have pathologic consequences. Particularly in more immature preterm infants, minute ventilation can diminish significantly during episodes of periodic breathing, and oxygen saturations can decrease to hypoxemic levels in association with longer apnea durations and increased time spent in periodic breathing. Since more immature infants generally spend more time in sleep and more of this sleep time is characterized by periodic breathing, these infants can spend a significant amount of time with low oxygen saturations. These extreme degrees of periodic breathing likely reflect a greater general immaturity of the central respiratory control centers; the more “severe” the periodic breathing, the more likely an infant is to demonstrate overt clinical symptoms related to AOP.
It is very likely that both periodic breathing and AOP arise from the same pathophysiology, i.e., inability of the central respiratory control centers to adequately modulate multiple afferent inputs and resulting inhibitory outputs to the muscles of respiratory control. AOP may simply reflect a more severe variant of this dysmodulation. Not all studies have linked periodic breathing with more prolonged apneas (41), but preterm infants that demonstrate frequent intervals of periodic breathing also demonstrate an increased incidence of AOP and hence a general immaturity of the respiratory control system. While periodic breathing can thus be a normal respiratory pattern in both term and preterm infants, the higher the percent sleep time spent in periodic breathing and the greater the length of apnea during apneic periods, the more likely it is that these intervals represent a clinically significant immaturity of the central respiratory system associated with “pathologic” disturbances in central respiratory control.
The prevalence of periodic breathing is not precisely known since most studies have been based on convenience samples not necessarily representative of asymptomatic as well as symptomatic infants. Differing definitions for periodic breathing are an additional concern. In preterm infants less than 1,000 grams, many authors have reported the prevalence to be 100% (1,41). In term infants, the prevalence has been reported to be lower, but still as high as 80% (42,43). The high initial prevalence in both term and preterm neonates diminishes over time, most significantly in the first 6 to 8 weeks of life.
CLINICAL DISORDERS IN CONTROL OF BREATHING
Apnea
Apnea is defined as the cessation of breathing for longer than 20 seconds, or a shorter duration in the presence of pallor, cyanosis (See Color Plate), or bradycardia (44). There are multiple possible etiologic factors leading to symptomatic apnea in the preterm and full-term infant (Tables 28-1 and 28-2). The occurrence of apneic episodes thus requires clinical and laboratory assessments to rule out conditions requiring definitive treatment. There are no systematic prevalence data for symptomatic apnea in full-term infants, but most occurrences will have an identifiable medical cause (Table 28-2). In preterm infants less than 1,500 grams at birth, approximately 70% will have at least one clinically observed episode of symptomatic apnea while in the neonatal intensive care unit (NICU), and about 20% of these infants will have a specific medical cause (Table 28-1). The other 80% of preterm infants with symptomatic apnea do not have a specific medical cause and by exclusion are then diagnosed as having AOP, the most important and prevalent “disorder” of respiratory control occurring in preterm infants.
TABLE 28-1 ETIOLOGY OF APNEA IN PRETERM INFANTS | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Apnea of Prematurity
Pathophysiology
The maturation of cardiorespiratory control and the clinical course of premature infants with AOP parallel each other and are consistent with the data reported in fetal animal studies. AOP is a direct consequence of immaturity of brainstem respiratory control centers. Both the presence
and severity progressively increase the lower the gestational age. This autonomic immaturity is not unique to respiratory control centers, however. Although there is no objective way to clinically quantify degree of immaturity for other brainstem autonomic control systems, brainstem auditory maturation can be quantified by serial measurements of brainstem conduction time from the auditory-evoked response (wave VI interval) (45). Both improved synaptic efficiency and myelination appear to be responsible for shortening of brainstem auditory conduction times with advancing gestational age. The brainstem auditory nuclei are located in close proximity to the cardiorespiratory centers, and there is a strong association between long brainstem conduction times for the auditory-evoked responses and clinical episodes of AOP. Since not all very-low-birth-weight preterm infants develop AOP and the severity varies even among affected infants of the same gestational age, other genetic and/or environmental risk factors are also likely to be important. Although no genetic, neurotransmitter, or neuropathologic studies have been performed in preterm infants with AOP, performance of such studies may be as informative in AOP as they appear to be in other control-of-breathing disorders.
and severity progressively increase the lower the gestational age. This autonomic immaturity is not unique to respiratory control centers, however. Although there is no objective way to clinically quantify degree of immaturity for other brainstem autonomic control systems, brainstem auditory maturation can be quantified by serial measurements of brainstem conduction time from the auditory-evoked response (wave VI interval) (45). Both improved synaptic efficiency and myelination appear to be responsible for shortening of brainstem auditory conduction times with advancing gestational age. The brainstem auditory nuclei are located in close proximity to the cardiorespiratory centers, and there is a strong association between long brainstem conduction times for the auditory-evoked responses and clinical episodes of AOP. Since not all very-low-birth-weight preterm infants develop AOP and the severity varies even among affected infants of the same gestational age, other genetic and/or environmental risk factors are also likely to be important. Although no genetic, neurotransmitter, or neuropathologic studies have been performed in preterm infants with AOP, performance of such studies may be as informative in AOP as they appear to be in other control-of-breathing disorders.
TABLE 28-2 ETIOLOGY OF APNEA AT BIRTH IN FULL TERM INFANTS | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Apneic episodes in AOP are subclassified as central, obstructive, or mixed (34,46). Figure 28-3 demonstrates the respiratory patterns during these events. Central apneas result from lack of respiratory effort. Obstructive apneas (obstructed breaths) are also central in origin but are related to absence of neuromuscular control of upper airway patency rather than absence of inspiratory diaphragmatic stimulation. Hence, obstructive apneas are characterized
by cessation of inspiratory air flow into the lungs despite persisting respiratory effort. Mixed apneas represent a combination of missed breaths (central apnea) and obstructed breaths. Mixed episodes of AOP can begin either with obstructed breaths or with central apnea, and there can be multiple alternations between obstructed breaths and central apnea within any single episode. In terms of relative frequency of each type, mixed apnea generally accounts for 53% to 71% of events, with obstructive apnea 12% to 20% and central apnea 10% to 25% (34). Preterm infants with AOP have significantly lower ventilatory responses to CO2 than preterm infants matched for gestational age without AOP (31) (Fig. 28-2).
by cessation of inspiratory air flow into the lungs despite persisting respiratory effort. Mixed apneas represent a combination of missed breaths (central apnea) and obstructed breaths. Mixed episodes of AOP can begin either with obstructed breaths or with central apnea, and there can be multiple alternations between obstructed breaths and central apnea within any single episode. In terms of relative frequency of each type, mixed apnea generally accounts for 53% to 71% of events, with obstructive apnea 12% to 20% and central apnea 10% to 25% (34). Preterm infants with AOP have significantly lower ventilatory responses to CO2 than preterm infants matched for gestational age without AOP (31) (Fig. 28-2).
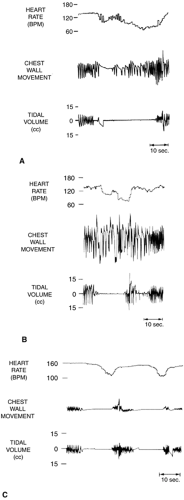 Figure 28-3 Examples of mixed, obstructive, and central apnea episodes occurring in apnea of prematurity. A: Mixed apnea. Obstructed breaths precede and follow a central respiratory pause. B: Obstructive apnea. Breathing efforts continue, although no nasal airflow occurs. C: Central apnea. Both nasal airflow and breathing efforts are absent. BPM, beats per minute. (Reprinted from Miller MJ, Martin RJ, Carlo WA. Diagnostic methods and clinical disorders in children. In Edelman NH, Santiago TV, eds. Breathing disorders of sleep. New York: Churchill Livingstone, 1986:157-180, with permission.) |
Incidence and Diagnosis
Threshold criteria for diagnosis have not been objectively defined and there is no established diagnostic test. Reported incidence data are therefore not based on any standardized criteria for diagnosis, but range from a low of less than 10% in preterm infants born at 34 weeks of gestation or more to approximately 60% at birth weights less than 1,500 grams and to a high of greater than 85% among infants born at less than 28 weeks of gestation (44,47,48). The first episodes can manifest themselves within the first 24 hours of life in spontaneously breathing infants without respiratory distress but are typically delayed in infants still receiving mechanical ventilatory support (49).
The identification of individual episodes of AOP is routinely based on detection of: a) central apnea lasting at least 20 seconds using standard impedance-based cardiorespiratory monitoring, b) bradycardia of less than 80 to 100 beats per minute (bpm) depending on gestational age, or c) arterial oxygen desaturation using a pulse oximeter (SpO2). These symptoms are typically associated with clinically observed cyanosis. To be certain that monitor alarms are clinically significant, episodes initially detected by a monitor alarm need to be confirmed by visual observation (34,50). Since impedance-based cardiorespiratory monitors cannot detect obstructive apneas unless and until associated with bradycardia, however, episodes of AOP will not be detected by the monitor until central apnea has persisted long enough to trigger a monitor alarm or until the bradycardia threshold has been reached. Isolated respiratory pauses of less than 20 seconds without associated bradycardia or desaturation may not be of clinical significance and are not sufficient to justify a diagnosis of AOP. Periodic breathing is typically prominent in infants with AOP but is not sufficient for diagnosis in the absence of associated bradycardia and/or desaturation.
In infants with AOP, the periodic respiratory pauses associated with periodic breathing may be longer than in infants without AOP, and the total time spent in periodic breathing may be longer. Both of these factors contribute to the occurrence and severity of associated bradycardia and/or desaturation (39). Visually confirmed episodes of apnea with associated heart rate decelerations and desaturation are classified as symptomatic episodes. There is no consensus-based threshold number of clinical episodes required for a “diagnosis” of AOP, but the minimum is at least one visually confirmed symptomatic episode unrelated to another medical cause (Table 28-1). The severity of AOP in individual patients and the threshold for initiating treatment are determined by the frequency and severity of subsequent clinically observed episodes. Although a multihour and multichannel recording that includes monitoring for both central and obstructive apneas, bradycardia, and desaturation might provide more objective threshold criteria for diagnosis and treatment, such recordings are not part of routine clinical care and no criteria have been established.
Treatment
Once other medical causes of apnea have been excluded (Table 28-1) and a diagnosis of AOP established, multiple treatment strategies are available. The first step is to ensure that borderline levels of baseline hypoxemia, anemia, hypocalcemia, and hypoglycemia have been corrected. Infants with intermittent hypoxemia due to episodes of AOP will be at increased risk for reduced tissue oxygen delivery (if they are also anemic) and for further exacerbation of AOP symptoms. Blood transfusions can be effective in reducing the severity of episodes (52,53), but some studies have not shown any correlation between degree of hypoxemia and degree of anemia (54,55 and 56). Some studies suggest that transfusion may be helpful in ameliorating symptoms of AOP only if the baseline hematocrit is less than 25 %. Clinicians should address the following questions when considering transfusion to improve symptoms related to AOP (57):
Are the symptoms related to AOP new in origin?
Has the severity of AOP increased as anemia has worsened?
Is the child receiving oxygen or other ventilatory support?
Has the heart rate increased?
Are other respiratory rhythm changes, such as periodic breathing, more prominent?
Affirmative answers to these questions increase the likelihood that a transfusion may improve clinical symptoms attributed to AOP.
The criteria for pharmacologic or ventilatory support vary among neonatologists, and there are no established clinical guidelines. Treatment is indicated, however, whenever clinical episodes are recurring, do not resolve spontaneously or in response to minimal stimulation, and are associated with bradycardia and intermittent hypoxemia. The first line of therapy is generally a methylxanthine, either caffeine or aminophylline.
Methylxanthines
Methylxanthines are central respiratory stimulants that increase CO2 sensitivity and hence lead to improved tidal
and minute volumes and blood gas values. Methylxanthines also increase diaphragmatic function, decrease muscle fatigue, and increase metabolic rate and catecholamine activity. These medications can be given by either oral or intravenous route. Both aminophylline and caffeine have been studied in placebo-controlled trials, and both have been shown to reduce the incidence of apnea (44,58,59 and 60). Table 28-3 reviews the recommended starting doses and dosing intervals for caffeine and aminophylline. The recommended therapeutic blood levels are 5 to 10 μg/mL for aminophylline and 8 to 20 μg/mL for caffeine. Methylxan-thine side effects are secondary to catecholamine stimulation and include tachycardia, jitteriness, and irritability. Caffeine has a wider therapeutic index and side effects are uncommon at the recommended blood levels. There is consequently less need to check frequent blood levels. Direct comparisons of aminophylline and caffeine indicate similar efficacy at equivalent levels of central ventilatory stimulation. At equivalent degrees of central chemostimulation, however, side effects are less with caffeine than with aminophylline (61,62,63). Many neonatologists thus recommend caffeine as the preferred methylxanthine for treatment of AOP.
and minute volumes and blood gas values. Methylxanthines also increase diaphragmatic function, decrease muscle fatigue, and increase metabolic rate and catecholamine activity. These medications can be given by either oral or intravenous route. Both aminophylline and caffeine have been studied in placebo-controlled trials, and both have been shown to reduce the incidence of apnea (44,58,59 and 60). Table 28-3 reviews the recommended starting doses and dosing intervals for caffeine and aminophylline. The recommended therapeutic blood levels are 5 to 10 μg/mL for aminophylline and 8 to 20 μg/mL for caffeine. Methylxan-thine side effects are secondary to catecholamine stimulation and include tachycardia, jitteriness, and irritability. Caffeine has a wider therapeutic index and side effects are uncommon at the recommended blood levels. There is consequently less need to check frequent blood levels. Direct comparisons of aminophylline and caffeine indicate similar efficacy at equivalent levels of central ventilatory stimulation. At equivalent degrees of central chemostimulation, however, side effects are less with caffeine than with aminophylline (61,62,63). Many neonatologists thus recommend caffeine as the preferred methylxanthine for treatment of AOP.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


