2. Broad based appendage
3. Receives coronary sinus
4. Connects to TV
2. Sagittal
3. Four chamber
4. Four chamber
2. Finger-like long narrow-based appendage
3. Flap valve of FO floats inside
4. Connects to MV
2. Parasternal short axis
3. Four chamber
4. Four chamber
2. TV attachment to septum
3. TV more apically positioned
4. Moderator band
5. Coarse trabeculation
6. Connects to a bifurcating pulmonary artery
2. Four chamber
3. Four chamber
4. Four chamber
5. Four chamber
6. Parasternal short axis
2. MV more basally positioned
3. MV attaches to the free wall only
4. Apex is wide open
5. Fine trabeculation
6. Connects to a non-bifurcating aorta with head and neck vessels arising from it
2. Four chamber
3. Parasternal long axis
4. Four chamber
5. Four chamber
6. Parasternal long axis
2. Bifurcates within first centimeter of its origin
3. Continues with DA into DAO
2. Parasternal short axis
3. Sagittal ductal arch
2. Branches out superiorly towards neck
3. In continuity with MV
2. Sagittal aortic arch view
3. Parasternal long axis
2. Trifurcates with LPA and RPA, and DA
3. In continuity with DA and DAO
2. Sagittal ductal arch
3. Sagittal ductal arch
2. Gives off head and neck vessels
3. Arises from the middle of chest
2. Sagittal aortic arch
3. Sagittal aortic arch
DA, ductus arteriosus; DAO, descending aorta; FO, foramen ovale; IVC, inferior vena cava; LPA, left pulmonary artery; MV, mitral valve; RPA, right pulmonary artery; SVC, superior vena cava; TV; tricuspid valve.
Therefore, methodical application of other multiple scanning views should be utilized for accurate detection of all congenital heart anomalies (Table 8.2). In addition, M-mode, color flow, and pulse wave Doppler modalities (as deemed appropriate) should be employed when assessing integrity and function of those structures[14,15]. Measurement of cardiac structures should be performed in systole and diastole (Table 8.3 and Figure 8.1), and their values should be compared with established normal ranges in the corresponding gestation (Table 8.4)[15–18].
| Scanning view | Cardiac anomalies |
|---|---|
| A. Four-chamber view | 1. Primum ASD 2. VSD 3. AVSD 4. Mitral or tricuspid atresia 5. Ebstein’s anomaly of the tricuspid valve 6. Enlargement of atriums and ventricles 7. Hypoplastic left or right ventricle 8. Atrial septal aneurysm 9. Double inlet left ventricle 10. Abnormal connection of pulmonary veins 11. Congenitally corrected TGA 12. Left superior vena cava to coronary sinus connection |
| B. Outflow tract views | |
| I. Left ventricular outflow tract (parasternal long axis) | 1. Aortic stenosis or atresia 2. TGA 3. Double outlet right ventricle 4. Truncus arteriosus 5. VSD 6. Left SVC to coronary sinus connection |
| II. Right ventricular outflow (parasternal short axis) | 1. Pulmonary stenosis or atresia 2. Absent pulmonary valve syndrome 3. TOF 4. VSD |
| C. Three-vessel and trachea view | 1. CoA 2. Vascular ring, double aortic arch or right aortic arch 3. Left SVC to coronary sinus connection 4. Pulmonary artery hypoplasia 5. Aortic hypoplasia 6. Ductal anomalies, abnormal, restricted or reversed ductal flow 7. TGA 8. TOF |
| D. Bicaval long-axis view | 1. Interrupted IVC 2. Vein of Galen 3. Abnormal connection of pulmonary veins 4. Absent ductus venosus 5. Sinus venosus ASD 6. Restrictive FO |
| E. Ductal and aortic arch view | 1. CoA 2. TGA 3. Hypoplasia of aorta or pulmonary artery 4. Interrupted aortic arch |
ASD, atrial septal defect; AVSD, atrioventricular septal defect; CoA, coarctation of the aorta; FO, foramen ovale; IVC, inferior vena cava; SVC, superior vena cava; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
AV; atrioventricular; FO, foramen ovale; MV, mitral valve; RA, right atrium.
E/A, early ventricular filling velocity/late ventricular filling velocity; FO, foramen ovale.
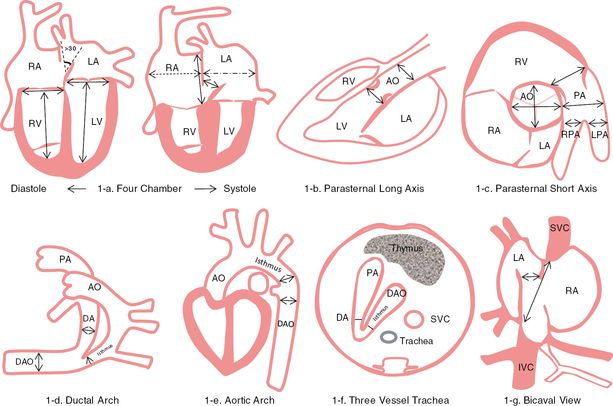
Measurement of cardiac structures. a) Measurement point of the four-chamber view in diastole and systole; b) from parasternal long axis view shows measurement of aortic valve and root; c) from parasternal short axis view shows aortic and pulmonary artery measurement points; d) from the ductal arch view shows ductus arteriosus, isthmus and descending aortic measurement points; e) from the aortic arch view shows isthmus and descending aortic measurement points; f) from the three vessel and trachea view shows measurement points of ductus arteriosus and the isthmus; g) from bicaval view shows measurement point of foramen ovale, IVC and SVC. LA, left atrium; RV, right ventricle; LV, left ventricle; AO, ascending aorta; PA, pulmonary artery; RPA, right pulmonary artery; LPA, left pulmonary artery; DA, ductus arteriosus; DV, ductus venosus; DAO, descending aorta; IVC, inferior vena cava; SVC, superior vena cava; FO, foramen ovale.
Ultrasound appearance of major cardiac anomalies and differential diagnosis
Normal fetal circulation
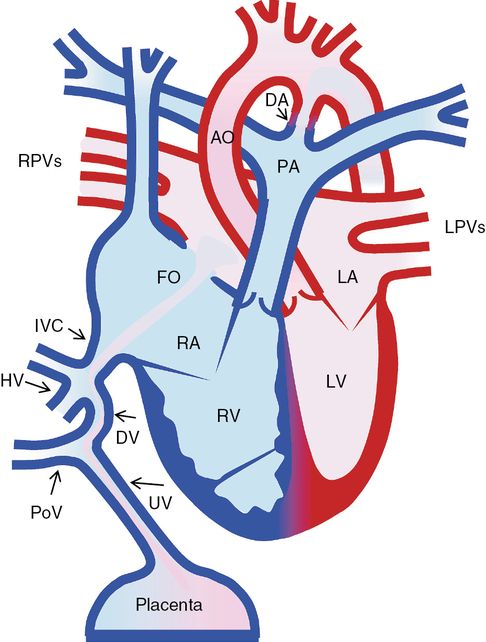
Normal fetal circulation. UV, umbilical vein; PoV, portal vein; DV, ductus venosus; HV, hepatic vein; IVC, inferior vena cava; RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; AO, ascending aorta; PA, pulmonary artery; DA, ductus arteriosus; DV, ductus venosus; DAO, descending aorta; IVC, inferior vena cava; RPVs, right pulmonary veins; LPVs, left pulmonary veins; FO, foramen ovale.
The placenta is the main blood supply for the fetus. Its main function is to provide oxygenation for fetal venous blood. There are three unique fetal cardiovascular connections: the ductus venosus (DV) (a connection between the umbilical vein and the inferior vena cava (IVC)), the foramen ovale (FO) (a communication between the right (RA) and the left atrium (LA)) and the ductus arteriosus (DA) (a connection between the pulmonary artery and the aorta) (Figure 8.2). Increased oxygen tension in the ductal tissue promotes closure of the DA within a few days after birth. As the pulmonary vascular resistance drops, pulmonary arterial blood flow increases significantly, which in turn leads to increased pulmonary venous return to the LA. Due to increased left atrial pressure, the flap valve of the FO moves rightward onto the septum secundum resulting in functional closure of the FO.
Outline of antenatal and postnatal management of major cardiac abnormalities
From the sonographic diagnostic point of view, it is more logical to describe heart defects by their diagnostic anatomic features in specific scanning views (Table 8.5). Congenital heart defects can also be classified according to their clinical importance as severe, moderately severe and minor anomalies (Table 8.6). Clinical classification of cardiac anomalies determines the need for prostaglandin infusion or requirement of surgery at birth or in the infancy, thereby allowing the fetal medicine specialist to decide place of delivery. Generally, half of congenital heart defects are major (severe) requiring surgery within the first year of life. There is also a subgroup of life-threatening severe congenital heart defects, so called “ductus arteriosus dependent congenital heart anomalies” (Table 8.7), which most certainly require delivery at a surgical center and infusion of prostaglandin-E immediately after birth. If such anomalies escape detection antenatally or after birth, they may lead to cardiovascular collapse and sudden death following closure of the DA. Between 35% and 65% of pregnancies with major (severe) cardiac anomalies are medically terminated (Table 8.5), which might affect the postnatal frequency of a specific congenital heart defect (Table 8.8) [19–24]. Antenatal and postnatal management of major congenital cardiac anomalies are summarized in Tables 8.6 to 8.9 [25–39]. We will now describe each cardiac anomaly and its diagnostic features and long-term outlook in the following section.
AVSD, atrioventricular septal defect; CCTGA; congenitally corrected transposition of the great arteries; DORV, double outlet right ventricle; IVC, inferior vena cava; LA, left atrium; LV, left ventricle; PA, pulmonary artery; PS, pulmonary stenosis; RV, right ventricle; SVC, superior vena cava; TGA, transposition of the great arteries; VSD, ventricular septal defect.
AS, aortic stenosis; AVSD, atrioventricular septal defect; CHD, congenital heart disease; CoA, coarctation of the aorta; DORV, double outlet right ventricle; FO, foramen ovale; PS, pulmonary stenosis; TA, tricuspid atresia; TAPVC, total anomalous pulmonary venous connection; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
| Cardiac anomaly | Antenatal management | Postnatal management |
|---|---|---|
| A. DA dependent systemic and pulmonary circulation 1. Transposition of the great arteries. B. DA dependent systemic circulation 1. Hypoplastic left heart syndrome 2. Critical aortic stenosis, 3. Aortic atresia 4. Coarctation of the aorta 5. Interrupted aortic arch C. Ductus arteriosus dependent pulmonary circulation 1. Critical pulmonary stenosis 2. Pulmonary atresia | The DA must remain open therefore avoid the use of ibuprofen, indomethacin, and discourage excess consumption of polyphenol rich food and drinks. The FO must remain open, hence monitor patency of the interatrial communication with bi-weekly ultrasound. Monitor pulmonary vein Doppler to help identify FO restriction in HLHS. Discuss at MDT meeting with neonatologist and pediatric cardiologist. Check karyotype to exclude 22q11 microdeletion in IAA, PS with VSD; Noonan syndrome in critical PS; Turner syndrome in coarctation; and William’s syndrome in supravalvar aortic stenosis. | Normal planned delivery with induction at 39 weeks in a surgical center if local facilities are away from a tertiary surgical center. Delivery at a nonsurgical center may be feasible if there is local pediatric cardiology service, local neonatal intensive care and neonatal transfer facilities are available. Prostaglandin infusion at 5–10 ng/kg/min should be started after birth. Avoid excess use of oxygen or unnecessary intubation. Oxygen saturations between 70–90% are acceptable. Nil by mouth until diagnosis is established and management plan is drawn |
CoA, coarctation of the aorta; DA, ductus arteriosus; FO, foramen ovale; HLHS, hypoplastic left heart syndrome; IAA, interrupted aortic arch; MDT, multidisciplinary team; TGA, transposition of the great arteries.
| Heart anomaly | Fetal frequency among CHD % | Incidence in per 1,000 live birth | Outcome |
|---|---|---|---|
| Atrioventricular septal defect | 5–16 | 0.19–0.35 | 1. <2% operative mortality 2. 95% survival at 20 year 3. 25% reoperation rate for AV valve regurgitation 4. 40% associated with Down’s syndrome, 20% trisomy 13 and 18 |
| Ventricular septal defect | 15–35 | 1.1–3.57 | 1.<2% operative mortality 2. Risk of complete heart block postoperatively 3. Excellent long term outlook |
| Hypoplastic left heart syndrome/ critical aortic stenosis | 4–16 | 0.14–0.27 | 1. 20% IU death 2. >85% survival after stage 1 Norwood and >98% survival after Glenn and Fontan 3. 50% survival in 10 years 4. 15% association with chromosome abnormalities, Turner most common 5. Isolated aortic stenosis 25-year survival >70% 6. Need for repeat operation to replace aortic valve |
| Tricuspid atresia | 1–3 | 0.03–0.5 | 1. >90% survival with arterial shunt, >98% survival with Glenn operation and >98% survival with Fontan operation. 2. 85% survival at 10 years 3. 8% associated with 22q11 deletion |
| Total anomalous pulmonary venous connection | 0.6–3 | 0.06–0.09 | 1. Excellent long term outlook 2. 2% risk of pulmonary vein stenosis |
| Ebstein’s anomaly | 0.3–0.7 | 0.06–0.11 | 1. IU mortality up to 45–100 % in severe cases, up to 90% survival in mild cases 2. May be associated with trisomy 21 |
| Double inlet left ventricle | 0.07–0.1 | N/A | 1. 10 year survival >70% |
| Tetralogy of Fallot | 3–7 | 0.42 | 1. 8–23% 22q11 deletion, up to 75% if absent pulmonary valve syndrome 2. Tetralogy has excellent long term survival but up to 30% reoperation for PS or regurgitation 3. 30–50% mortality in absent pulmonary valve syndrome |
| Truncus arteriosus | 3–4.8 | 0.05–0.11 | 1. 40% 22q11 deletion 2. 50% truncal valve regurgitation or stenosis 3. Early pre-operative death 5–10%, early survival 90% at 5 years |
| Double outlet right ventricle | 2.4–12 | 0.16 | 1. Survival 70–80% at medium term |
| Severe pulmonary stenosis or atresia | 2.4–6.5 | 0.16–0.26 | 1. PA-VSD 8–23% associated with 22q11 deletion 2. PS may be associated with Noonan’s, Alagille, and Leopard syndromes 3. Outlook is good in PS and tripartite right ventricle 4. PA-IVS survival 65% at 5 years and PA-VSD 70% survival at 10 years |
| Transposition of the great arteries | 4.3–11 | 0.24–0.32 | 1. Excellent long term survival 97% at 10 years 2. Branch pulmonary artery stenosis 5–30% |
| Coarctation/interruption of aorta | 8.9 | 0.14–0.41 | 1. 50% of interruption has 22q11 and 35% of Turner has coarctation 2. 25% associated with noncardiac anomalies 3. Excellent survival 4. 4% recurrence rate and reoperation risk, 25% risk of hypertension |
AV, atrioventricular; CHD, congenital heart disease; IU, intrauterine; PA-VSD, pulmonary artery-ventricular septal defect.
CoA, coarctation of the aorta; DA, ductus arteriosus; FO, foramen ovale; PS, pulmonary stenosis; SVC, superior vena cava; VSD, ventricular septal defect.
Intracardiac shunts that can be detected in four-chamber view
Ventricular septal defect
Ventricular septal defect (VSD) is a communication between the left and right ventricles (Figure 8.3). It is the most common of all congenital heart lesions and usually occurs as an isolated abnormality. It can also be associated with coarctation of the aorta (CoA), atrioventricular septal defect (AVSD), tetralogy of Fallot (TOF) and truncus arteriosus.
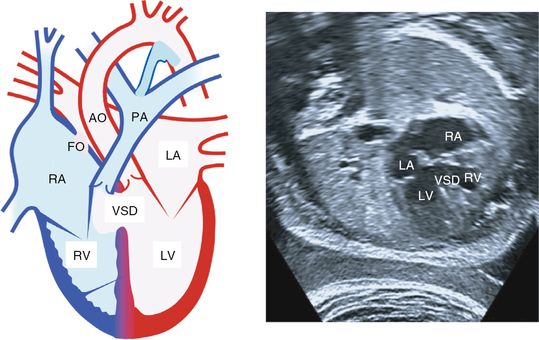
Ventricular septal defect. Left: from the four-chamber view shows a ventricular septal defect. Echocardiography on the right from the four-chamber view shows a ventricular septal defect. RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; VSD, ventricular septal defect; FO, foramen ovale; PA, pulmonary artery; AO, aorta.
There are three major locations: underneath the inlet valves (perimembranous-inlet), in the middle of ventricular septum (muscular) and underneath outflow tracts (outlet). Muscular and perimembranous-inlet type defects can be detected in the four-chamber view, but the five-chamber, long-axis and short views are all needed to demonstrate the outflow defects. Color flow or pulse wave Doppler may aid in diagnosing smaller defects by showing bidirectional flow pattern across the ventricular septum. False-positive and negative rates are high.
The fetal clinical course is uncomplicated. The majority of small VSDs close spontaneously either in utero or after birth. It is important to exclude commonly associated defects such as CoA, TOF and truncus arteriosus.
Amniocentesis should be recommended for large VSDs located in inlet or outlet septum, to exclude 22q11 microdeletion and trisomy 21[38]. Delivery can take place at a local hospital if the VSD is an isolated anomaly regardless of its size.
Postnatally, no special precaution is needed in the newborn other than routine pediatric and cardiac review. A large VSD may lead to faltering growth and heart failure in infancy.
Treatment involves either stitch or patch closure of the defect with surgery. Some anatomically suitable defects may be amenable to device closure via percutaneous transcatheter approach. VSD has an excellent late functional and survival outcome after treatment.
Complete atrioventricular septal defect
AVSD has an atrial and ventricular communication with a common atrioventricular (AV) valve (Figure 8.4). In the incomplete form, there is an ostium primum atrial septal defect and a cleft mitral valve without a defect in the ventricular septum. This abnormality can be suspected from the four-chamber view by showing the left and right inlet valves being at the same level instead of having a normal off-setting appearance.
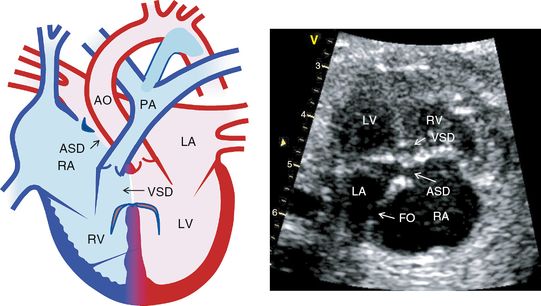
Atrioventricular septal defect. Diagram on the left from the four-chamber view shows atrioventricular septal defect. Echocardiography on the right from the four chamber view shows atrioventricular septal defect. RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle, ASD, atrial septal defect; VSD, ventricular septal defect; PA, pulmonary artery; AO, aorta.
Fetuses with this defect are commonly asymptomatic, but due to its frequent associations with chromosome abnormalities, amniocentesis is strongly recommended. Approximately 40–50% of fetuses with AVSD will have trisomy 21, 18 or 13. Likewise, 40–50% of fetuses with trisomy 21 will have AVSD. AVSD may also coexist with pulmonary stenosis in 2–10% of cases[36].
Postnatally, neonates with AVSD may be totally asymptomatic or only slightly cyanosed, and diagnosis in some cases may be overlooked. Any newborn with AVSD should have a careful pediatric, cardiac and if necessary, genetics review. Percutaneous oxygen saturation should be monitored after birth. Once the pulmonary vascular resistance drops, the neonates with a large VSD may develop respiratory symptoms, faltering growth and heart failure generally beyond 2 weeks of age.
Surgical repair of complete AVSD is usually undertaken before 6 months of age. After surgical repair, reasonable quality of life is expected, but there is a 20% re-operation rate for leaky inlet valves. However, if there is disproportionate ventricular size (unbalanced AVSD), the postnatal outlook is less unfavorable as the corrective two-ventricle repair cannot be achieved.
Anomalous pulmonary venous connection
In total anomalous pulmonary venous connection (TAPVC), some or all pulmonary veins fail to connect to the LA (Figure 8.5). Instead they may connect to the superior vena cava (SVC), RA, or into the liver either directly or indirectly via a common channel. An obstructed venous channel should be excluded. This abnormality is notoriously difficult to detect antenatally with no more than 6–10% detection rates. Presence of nonpulsatile pulmonary vein Doppler flow may alert the sonographer to this diagnosis.
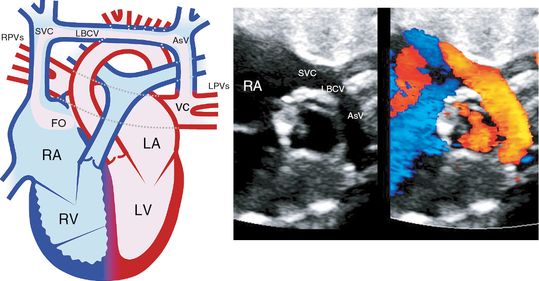
Total anomalous pulmonary venous connection. Diagram on the left from the four chamber view shows supracardiac total anomalous pulmonary venous connection to the left brachiocephalic vein. Echocardiography on the right from the four-chamber view shows total anomalous venous connection to the left brachiocephalic vein. RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; AV, ascending vein; LBCV, left brachiocephalic vein; SVC, superior vena cava; PV, pulmonary vein.
Fetuses with this abnormality are asymptomatic. TAPVC is rarely associated with chromosome abnormality. Usual prenatal follow-up at 4–6 weeks’ intervals is required.
Postnatally some of these patients may exhibit very little in the way of cyanosis, and diagnosis can easily be overlooked. If there is pulmonary venous obstruction, cyanosis will be accompanied by respiratory difficulties, which may frequently mimic persistent pulmonary hypertension of the newborn.
Surgical treatment involves re-implantation of pulmonary veins into the LA. Once corrected, most patients remain asymptomatic with no major long-term issues.
Outflow tract abnormalities
TOF and double outlet right ventricle
TOF has four features: severe narrowing of the right ventricular outflow (infundibular pulmonary stenosis), a large subaortic VSD, overriding of the aortic outlet above the ventricular septum and right ventricular hypertrophy (Figure 8.6). The 22q11 chromosome microdeletion is not infrequently (5–10%) associated with this anomaly. The four-chamber view may be completely normal, but an extended apical five-chamber view should demonstrate the VSD and the aortic override. The parasternal short-axis view is necessary to show the site of pulmonary stenosis.
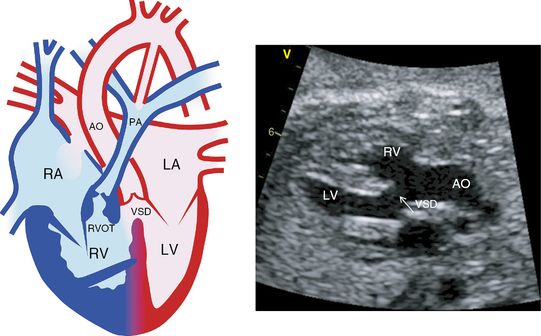
Tetralogy of Fallot. Diagram on the left from the four-chamber view shows right ventricular hypertrophy, right ventricular outflow tract obstruction, ventricular septal defect and an overriding aorta. On the left, echocardiography from the parasternal long axis view shows an overriding aorta and ventricular septal defect. This view does not demonstrate pulmonary stenosis which can be obtained from the parasternal short axis view. RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; RVOT, right ventricular outflow tract; VSD, ventricular septal defect; AO, aorta; PA, pulmonary artery.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


