Congenital Bronchopulmonary Malformations
Congenital bronchopulmonary malformations (BPMs) represent a continuum of abnormalities of the bronchopulmonary unit for which classification and management remain in evolution. An improved understanding of the molecular mechanisms underlying the embryologic development of the lung and the pathogenesis of BPMs suggests that these lesions may have similar mechanistic origins that differ in developmental timing or location in the bronchopulmonary tree.1 From a clinical perspective, improvements in prenatal imaging and increasing observational experience have resulted in a better understanding of the natural history of these anomalies, and a better predictive capacity for pre-, peri-, and postnatal events. Finally, postnatal treatment for the majority of lesions has improved with advances in neonatal care, and the development of thoracoscopic surgery. This chapter discusses the prenatal and postnatal management of the major congenital BPMs with an emphasis on utilizing the thoracoscopic approach for management.
Embryology and Development of the Bronchopulmonary Tree
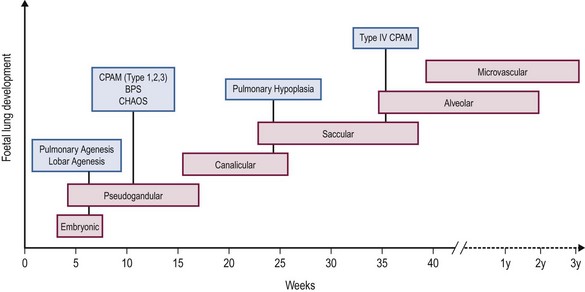
Embryological development of the human lung transitions through six separate stages to form a bronchial tree with greater than 1 × 105 conducting and 1 × 107 respiratory airways.2 These stages include embryonic, pseudoglandular, canalicular, saccular, alveolar, and microvascular. The progression of each stage is a highly coordinated process guided by mesenchymal–epithelial interactions under the influence of a number of regulatory growth factors.
Briefly, the embryonic phase of lung development begins with the formation of the laryngotracheal bud from the anterior portion of the primitive gut. Beginning at week 5 in the pseudoglandular phase, the preacinar airways and blood vessels develop, followed by growth of the bronchial tree until all bronchial divisions are completed by 16 weeks gestation.3 The cannalicular stage follows, and is characterized by capillary growth towards the respiratory epithelium which marks the future blood–air interface.2 The transition to the saccular stage at 24 weeks is marked by the widening of peripheral air spaces distal to the terminal bronchioles with septa formation. The final stages of lung development include the alveolar stage, defined by the formation of secondary septa and budding alveoli, and followed by the microvascular stage with significant alveolar development and maturation. During this complex process, the timing of congenital BPMs and their pathogenesis can be related to specific time points in each of the six developmental stages (Fig. 22-1).
Prenatal Diagnosis and Classification of Congenital Bronchopulmonary Malformations
Malformations
Prenatal diagnosis and fetal therapy for congenital lung malformations have evolved significantly since Adzick et al. described the near universal mortality of congenital pulmonary airway malformation (CPAM)-induced fetal hydrops almost three decades ago.4 Congenital BPMs represent 90% of lung lesions seen in clinical practice and include CPAMs, (formally called congenital cystic adenomatoid malformation or CCAM), bronchopulmonary sequestration (BPS), and congenital lobar emphysema (CLE).5 Other less common malformations are varied and are included in the classification system described by Langston et al.6 (Box 22-1), but will not be discussed in this chapter.
Prenatal ultrasonography (US) functions as a window into fetal development and is the most common mode of prenatal diagnosis of congenital thoracic abnormalities (Box 22-2). We routinely supplement ultrasound with magnetic resonance imaging (MRI) as a complementary method to further define the anatomy of the lesion and overall fetal morphology.7,8 In combination, the two modalities allow accurate prenatal diagnosis of the different types of BPMs and exclude other anatomic anomalies.
Congenital Pulmonary Airway Malformation
CPAMs are the most commonly diagnosed BPM. The best estimate of the incidence of CPAM is 0.66 per 10,000 live births, but better studies are needed based on high quality prenatal imaging of all pregnancies in a study population.9 This heterogeneous group of congenital cystic and noncystic lung masses is characterized by an extensive overgrowth of immature primary bronchioles localized to a segment of the bronchial tree (Fig. 22-2).10 The current classification system defined by Stocker classifies CPAMs into five types that differ by location, cystic structure, size, and epithelial lining (Table 22-1). However, from a practical perspective, the prenatal classification of CPAMs is divided into two categories based on prenatal ultrasound findings: (1) macrocystic lesions containing a single or multiple cysts that are 5.0 mm in diameter or greater; and (2) microcystic lesions presenting as a solid echogenic mass on prenatal ultrasound (Fig. 22-3).4
TABLE 22-1
Stocker Classification: Congenital Pulmonary Airway Malformations
From Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol 1977;8:155–71.
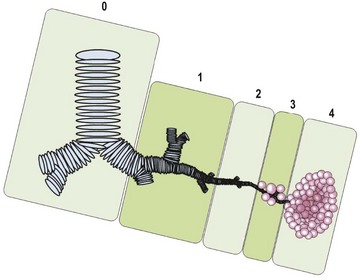
FIGURE 22-2 This CPAM classification schematic is based on the location of the development of the malformation. Type 0, tracheobronchial; 1, bronchial/bronchiolar; 2, bronchiolar; 3, bronchiolar/alveolar; 4, distal acinar. Adapted from Stocker (2009).63
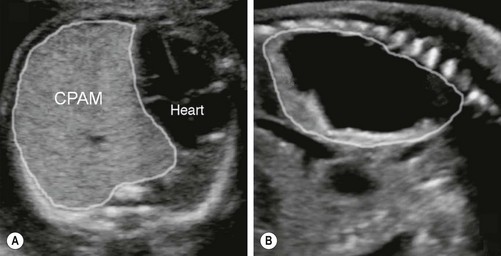
FIGURE 22-3 These two prenatal ultrasounds depict a practical prenatal classification for congenital pulmonary airway malformations (CPAM). (A) The prenatal ultrasound study finds microcystic lesions presenting as a solid echogenic mass. (B) macrocystic lesions contain either a large cyst or multiple cysts that are greater than 5.0 mm diameter.
In the prenatal period, ultrasound will usually demonstrate an area of hyperechogenic tissue with or without hypoechoic cysts that vary in number and size. Signs of mass effect may be seen, such as mediastinal shift, diaphragmatic eversion, and polyhydramnios. With very large lesions, heart failure (hydrops) due to mediastinal shift and cardiac compression can occur.11 CPAMs receive their blood supply from the pulmonary artery and have pulmonary venous drainage. However, there is a subset of CPAMs, known as hybrid lesions, where the blood supply also includes an anomalous systemic artery.12 These lesions demonstrate anatomic and histologic features of both CPAMs and BPS.
The diagnosis of CPAMs by an experienced sonographer is usually straightforward. However, there are specific entities that are commonly misdiagnosed (Table 22-2). A skilled sonographer can usually easily differentiate these lesions by understanding the blood supply (congenital diaphragmatic hernia [CDH], lung agenesis, BPS), observation of bowel peristalsis (CDH), documentation of the absence of one lung (lung agenesis), or visualization of bronchial dilation (bronchial atresia, congenital high airway obstruction syndrome [CHAOS]). MRI is also a useful adjunct for differentiating these entities. In our opinion, it should be applied routinely in fetal diagnostic centers.
TABLE 22-2
Pitfalls in Ultrasonography in the Diagnosis of CPAM
| Anomaly | Misdiagnosis |
| Right-sided congenital diaphragmatic hernia | Large right sided microcystic CPAM: similar echogenicity of liver to microcystic CPAM |
| Lung agenesis | Large microcystic CPAM: appearance of mediastinal shift with a large echogenic lung |
| Congenital high airway obstruction syndrome | Bilateral large microcystic CPAM: bilateral large echogenic lungs with diaphragmatic eversion |
| Main stem bronchial, lobar, or segmental atresia | Microcystic CCAM: hyperplasia of distal lung and increased echogenicity |
Bronchopulmonary Sequestration
Bronchopulmonary sequestrations comprise approximately 10% of prenatally diagnosed BPMs, and are characterized by a portion of the lung that does not connect to the tracheobronchial tree. These lesions have a systemic arterial supply that can arise from the aorta or various systemic arterial branches above or below the diaphragm, and may have systemic or pulmonary venous return. Two different types of sequestration are described: intralobar (ILS) and extralobar (ELS) which differ in their prenatal and postnatal characteristics. An ILS shares visceral pleural investment with normal lung and drains into the pulmonary venous system while an ELS has a separate pleural investment and may have either systemic or pulmonary venous drainage.13,14
ELS is seen as a homogeneous hyperechoic mass in a paraspinal location, most often in the left lower thorax (Fig. 22-4). The pathogenesis is related to a supernumerary lobe developing from abnormal budding early in foregut embryogenesis.15 If the bud arises before the development of the pleura, it is invested with the adjacent lung and becomes an ILS. If the bud develops after visceral pleural formation, it grows separately and acquires its own pleural covering.14 It is important to appreciate that ELS can be found at any level in the pleural space and are also found within or beneath the diaphragm (see Fig. 22-4B).15 The major feature that helps discriminate an ELS from a CPAM is the blood supply derived from the systemic circulation with systemic venous drainage identified by Doppler ultrasound or MRI. However, it is important to appreciate that some anatomic ELSs can contain visible cysts that ultimately are shown to have CPAM histology (Fig. 22-5). In addition, an ELS can occasionally have venous drainage via a large venous channel draining directly into a pulmonary vein that is usually identified as aberrant venous drainage by imaging studies.
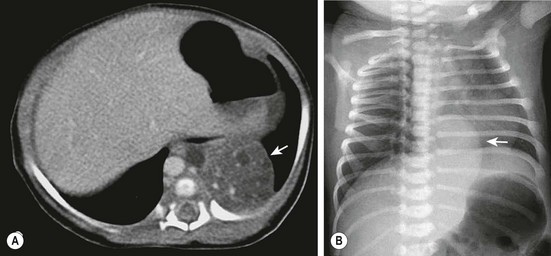
FIGURE 22-4 (A) The CT scan demonstrates an extralobar sequestration in the typical basilar location of the left chest (arrow). (B) In a different patient, the chest radiograph shows a large transdiaphragmatic extralobar sequestration (arrow).
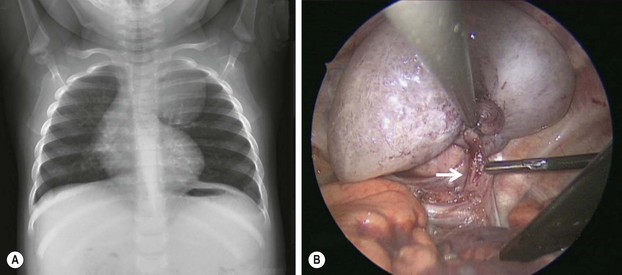
FIGURE 22-5 This 9-month-old developed an upper respiratory infection and a chest radiograph was performed. (A) The chest radiograph shows a left apical mediastinal mass. (B) After her infection resolved, she was found to have this extrapulmonary mass at thoracoscopy, along with a feeding vessel (arrow). The vessel was ligated and the mass removed, and she recovered uneventfully. Histologic examination showed the mass to an extralobar sequestration associated with a microcystic congenital pulmonary airway malformation.
Congenital Lobar Emphysema
CLE is a condition characterized by overinflation and distension of one or more pulmonary lobes with compression of the adjacent lung. In 50% of cases, the cause is unknown. In the remaining 50%, it may result from dysplastic bronchial cartilage, endobronchial obstruction, extrinsic compression from aberrant cardiopulmonary vasculature, or diffuse bronchial abnormalities related to infection.16 In the fetus, amniotic fluid trapping is analogous to air trapping and can lead to lobar expansion. The left upper lobe is the most frequently affected, followed by right middle and upper lobes, with rare bilateral or multifocal involvement. CLE is most commonly diagnosed in the neonate or infant presenting with respiratory distress.
Prenatal discrimination between CLE and CPAM or bronchial atresia may be difficult, but the absence of a systemic vascular supply differentiates this lesion from an ELS. Although complications such as polyhydramnios and hydrops have not been reported with CLE, perinatal respiratory distress correlates with the prenatal size of the lesion as manifest by mediastinal shift or compression of adjacent lung parenchyma.17
Bronchogenic Cysts and Bronchial Atresia/Stenosis
Bronchogenic cysts develop from abnormal budding of the tracheal diverticulum or the ventral aspect of the primitive foregut, which is not followed by bronchial development or branching. The result is a cavity that may or may not communicate with the airway and can be found in a variety of locations depending on the location of abnormal budding during foregut development. Histologically, these lesions are thin walled and have a bronchial epithelial lining, and are filled with mucus.18 Prenatal diagnosis of these lesions is usually made by ultrasound where they may be seen as an isolated cystic structure in the mediastinum, or causing bronchial obstruction with findings of bronchial dilation and lung hyperplasia distal to the point of obstruction. Bronchial atresia without a bronchogenic cyst also results in hyperplasia distal to the level of obstruction and is frequently associated with mucocele formation. The presence of dilated bronchi indicates a diagnosis of atresia rather than microcystic CPAM. The more proximal the atresia, the greater the potential for mass effect manifest by mediastinal shift, and ultimately, fetal hydrops. Segmental bronchial stenosis/atresia is a relatively recently recognized abnormality characterized by an echogenic segment of lung on ultrasound that is indistinguishable from and usually diagnosed as a microcystic CPAM.19
Prenatal and Perinatal Management of Bronchopulmonary Malformations
Congenital Pulmonary Airway Malformation
Experience with serial imaging of large numbers of fetuses with CPAMs has clarified the pre- and perinatal natural history of this anomaly. There is a typical pattern of growth of a CPAM with a period of growth relative to the size of the fetus until approximately 26 weeks gestation at which time growth plateaus. After 28 weeks, the CPAM typically gets smaller relative to the size of the fetus as measured by the CPAM volume ratio or CVR. The CVR is calculated by dividing the volume of the CPAM (length × height × width × 0.52) by the head circumference. In addition, the CVR has proven on retrospective and prospective assessment to be the most useful predictor for the development of hydrops.20
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


