Chapter 585 Congenital Anomalies of the Central Nervous System
585.1 Neural Tube Defects
Stephen L. Kinsman and Michael V. Johnston
The human nervous system originates from the primitive ectoderm that also develops into the epidermis. The ectoderm, endoderm, and mesoderm form the three primary germ layers that are developed by the 3rd wk. The endoderm, particularly the notochordal plate and the intraembryonic mesoderm, induces the overlying ectoderm to develop the neural plate in the 3rd wk of development (Fig. 585-1A). Failure of normal induction is responsible for most of the NTDs, as well as disorders of prosencephalic development. Rapid growth of cells within the neural plate causes further invagination of the neural groove and differentiation of a conglomerate of cells, the neural crest, which migrate laterally on the surface of the neural tube (see Fig. 585-1B). The notochordal plate becomes the centrally placed notochord, which acts as a foundation around which the vertebral column ultimately develops. With formation of the vertebral column, the notochord undergoes involution and becomes the nucleus pulposus of the intervertebral disks. The neural crest cells differentiate to form the peripheral nervous system, including the spinal and autonomic ganglia and the ganglia of cranial nerves V, VII, VIII, IX, and X. In addition, the neural crest forms the leptomeninges, as well as Schwann cells, which are responsible for myelination of the peripheral nervous system. The dura is thought to arise from the paraxial mesoderm. In the region of the embryo destined to become the head, similar patterns exist. In this region, the notocord is replaced by the precordal mesoderm.
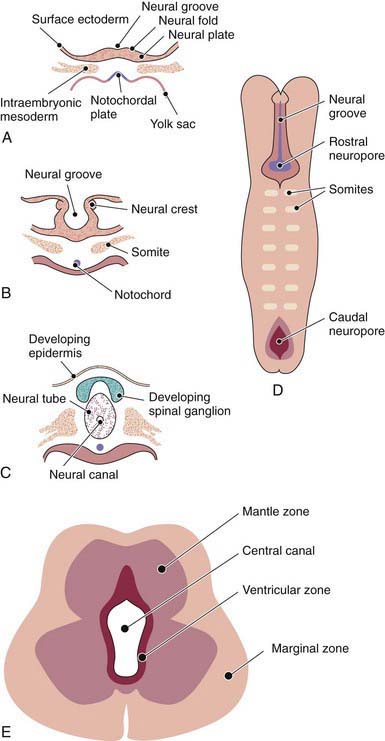
In the 3rd wk of embryonic development, invagination of the neural groove is completed and the neural tube is formed by separation from the overlying surface ectoderm (see Fig. 585-1C). Initial closure of the neural tube is accomplished in the area corresponding to the future junction of the spinal cord and medulla and moves rapidly both caudally and rostrally. For a brief period, the neural tube is open at both ends, and the neural canal communicates freely with the amniotic cavity (see Fig. 585-1D). Failure of closure of the neural tube allows excretion of fetal substances (α-fetoprotein [AFP], acetylcholinesterase) into the amniotic fluid, serving as biochemical markers for a NTD. Prenatal screening of maternal serum for AFP in the 16th-18th wk of gestation is an effective method for identifying pregnancies at risk for fetuses with NTDs in utero. Normally, the rostral end of the neural tube closes on the 23rd day and the caudal neuropore closes by a process of secondary neurulation by the 27th day of development, before the time that many women realize they are pregnant.
The embryonic neural tube consists of three zones: ventricular, mantle, and marginal (see Fig. 585-1E). The ependymal layer consists of pluripotential, pseudostratified, columnar neuroepithelial cells. Specific neuroepithelial cells differentiate into primitive neurons or neuroblasts that form the mantle layer. The marginal zone is formed from cells in the outer layer of the neuroepithelium, which ultimately becomes the white matter. Glioblasts, which act as the primitive supportive cells of the CNS, also arise from the neuroepithelial cells in the ependymal zone. They migrate to the mantle and marginal zones and become future astrocytes and oligodendrocytes. The importance of other pathways of progenitor cell generation and migration are also being elucidated. It is likely that microglia originate from mesenchymal cells at a later stage of fetal development when blood vessels begin to penetrate the developing nervous system.
585.2 Spina Bifida Occulta (Occult Spinal Dysraphism)
Stephen L. Kinsman and Michael V. Johnston
Spina bifida occulta is a common anomaly consisting of a midline defect of the vertebral bodies without protrusion of the spinal cord or meninges. Most patients are asymptomatic and lack neurologic signs, and the condition is usually of no consequence. Some consider the term spina bifida occulta to denote merely a posterior vertebral body fusion defect. This simple defect does not have an associated spinal cord malformation. Other clinically more significant forms of this closed spinal cord malformation are more correctly termed occult spinal dysraphism. In most of these cases, there are cutaneous manifestations such as a hemangioma, discoloration of the skin, pit, lump, dermal sinus, or hairy patch (Fig. 585-2). A spine roentgenogram in simple spina bifida occulta shows a defect in closure of the posterior vertebral arches and laminae, typically involving L5 and S1; there is no abnormality of the meninges, spinal cord, or nerve roots. Occult spinal dysraphism is often associated with more significant developmental abnormalities of the spinal cord, including syringomyelia, diastematomyelia, and/or a tethered cord. A spine roentgenogram in these cases might show bone defects or may be normal. All cases of occult spinal dysraphism are best investigated with MRI (Fig. 585-3). Initial screening in the neonate may include ultrasonography.
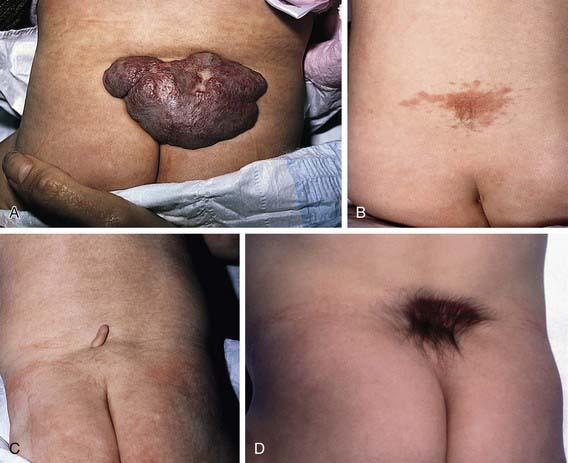
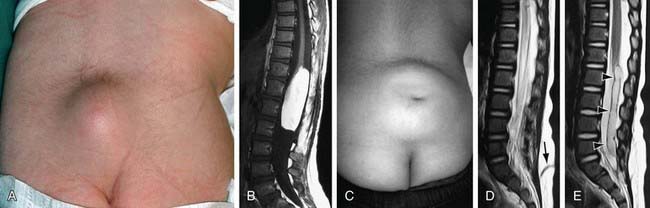
An approach to imaging of the spine in patients with cutaneous lesions is noted in Table 585-1.
Table 585-1 CUTANEOUS LESIONS ASSOCIATED WITH OCCULT SPINAL DYSRAPHISM
IMAGING INDICATED
IMAGING UNCERTAIN
IMAGING NOT REQUIRED
From Williams H: Spinal sinuses, dimples, pits and patches: what lies beneath? Arch Dis Child Educ Pract Ed 91:ep75–ep80, 2006.
585.4 Myelomeningocele
Clinical Manifestations
Myelomeningocele produces dysfunction of many organs and structures, including the skeleton, skin, and gastrointestinal and genitourinary tracts, in addition to the peripheral nervous system and the CNS. A myelomeningocele may be located anywhere along the neuraxis, but the lumbosacral region accounts for at least 75% of the cases. The extent and degree of the neurologic deficit depend on the location of the myelomeningocele and the associated lesions. A lesion in the low sacral region causes bowel and bladder incontinence associated with anesthesia in the perineal area but with no impairment of motor function. Newborns with a defect in the midlumbar or high lumbothoracic region typically have either a saclike cystic structure covered by a thin layer of partially epithelialized tissue (Fig. 585-4) or an exposed flat neural placode without overlying tissues. When a cyst or membrane is present, remnants of neural tissue are visible beneath the membrane, which occasionally ruptures and leaks CSF, whereas the placode is composed of neural tissue.
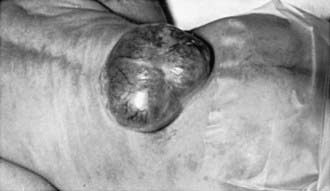
Treatment
Although incontinence of fecal matter is common and is socially unacceptable during the school years, it does not pose the same organ-damaging risks as urinary dysfunction, but occasionally fecal impaction and/or megacolon develop. Many children can be bowel-trained with a regimen of timed enemas or suppositories that allows evacuation at a predetermined time once or twice a day. Special attention to low anorectal tone and enema administration and retention is often required. Appendicostomy for antegrade enemas may also be helpful (Chapter 21.4).
In utero surgical closure of a spinal lesion has been successful in a few centers. Preliminary reports suggest a lower incidence of hindbrain abnormalities and hydrocephalus (fewer shunts) as well as improved motor outcomes. This suggests that the defects may be progressive in utero and that prenatal closure might prevent the development of further loss of function. In utero diagnosis is facilitated by maternal serum α-fetoprotein screening and by fetal ultrasonography (Chapter 90).
Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364:993-1004.
Bauer SB. Neurogenic bladder: etiology and assessment. Pediatr Nephrol. 2008;23:541-551.
Beeker T, Scheers M, Faber J, et al. Prediction of independence and intelligence at birth on meningomyelocele. Childs New Syst. 2006;22:33-37.
Bitsko RH, Reefhuis J, Romitti PA, et al. Periconceptional consumption of vitamins containing folic acid and risk for multiple congenital anomalies. Am J Med Genet. 2007;143A:2397-2405.
Bol KA, Collins JS, Kirby RS. Survival of infants with neural tube defects in the presence of folic acid fortification. Pediatrics. 2006;117:803-813.
Cameron M, Moran P. Prenatal screening and diagnosis of neural tube defects. Prenat Diagn. 2009;29:402-411.
Centers for Disease Control and Prevention. CDC Grand Rounds: additional opportunities to prevent neural tube defects with folic acid fortification. MMWR Morb Mortal Wkly Rep. 2010;59:980-984.
Chescheir NC. Maternal-fetal surgery: where are we and how did we get here? Obstet Gynecol. 2009;113:717-731.
Cochrane DD. Cord untethering for lipomyelomeningocele: expectation after surgery. Neurosurg Focus. 2007;23:1-7.
de Jong TP, Chrzan R, Klijn AJ, et al. Treatment of the neurogenic bladder in spina bifida. Pediatr Nephrol. 2008;23:889-896.
Dicianno BE, Kurowski BG, Yang JM, et al. Rehabilitation and medical management of the adult with spina bifida. Am J Phys Med Rehabil. 2008;87:1027-1050.
Fichter MA, Dornseifer U, Henke J, et al. Fetal spina bifida repair—current trends and prospects of intrauterine neurosurgery. Fetal Diagn Ther. 2008;23:271-286.
Guggisberg D, Hadj-Rabia S, Viney C, et al. Skin markers of occult spinal dysraphism in children. Arch Dermatol. 2004;140:1109-1115.
Ickowicz V, Ewin D, Maugay-Laulom B, et al. Meckel-Gruber syndrome, sonography and pathology. Ultrasound Obstet Gynecol. 2006;27:296-300.
Joseph DB. Current approaches to the urologic care of children with spina bifida. Curr Urol Rep. 2008;9:151-157.
Kibar Z, Torban E, McDearmid JR, et al. Mutations in VANGLI associated with neural-tube defects. N Engl J Med. 2007;356:1432-1437.
Scales CD, Wiener JS. Evaluating outcomes of enterocystoplasty in patients with spina bifida: a review of the literature. J Urol. 2008;180:2323-2329.
Stevenson RE, Allen WP, Pai GS, et al. Decline in prevalence of neural tube defects in a high-risk region of the United States. Pediatrics. 2000;106:677-683.
Tubbs RS, Bui CJ, Loukas M, et al. The horizontal sacrum as an indicator of the tethered spinal cord in spina bifida aperta and occulta. Neurosurg Focus. 2007;23:1-4.
U.S. Preventive Services Task Force. Recommendation statement: folic acid for the prevention of neural tube defects. Ann Intern Med. 2009;150:626-631.
Williams H. Spinal sinuses, dimples, pits and patches: what lies beneath? Arch Dis Child. 2006;91:ep75-ep80.
585.7 Disorders of Neuronal Migration
Disorders of neuronal migration can result in minor abnormalities with little or no clinical consequence (small heterotopia of neurons) or devastating abnormalities of CNS structure and/or function (mental retardation, seizures, lissencephaly, and schizencephaly, particularly the open-lip form) (Fig. 585-5). One of the most important mechanisms in the control of neuronal migration is the radial glial fiber system that guides neurons to their proper site. Migrating neurons attach to the radial glial fiber and then disembark at predetermined sites to form, ultimately, the precisely designed six-layered cerebral cortex. Another important mechanism is the tangential migration of progenitor neurons destined to become cortical interneurons. The severity and the extent of the disorder are related to numerous factors, including the timing of a particular insult and a host of environmental and genetic contributors.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


