Congenital Anomalies
Scott Douglas McLean
Much of our practice of pediatrics and neonatology is geared toward the provision of acute care, such as resuscitation, antibiotics, and intravenous fluids. When we encounter a newborn with one or more birth defects, this acute-care paradigm remains—and appropriately so—in the forefront of our clinical perspective. However, the child with a congenital anomaly must also be simultaneously considered from a different perspective, one that looks far back and far forward in time. When did this structuralnormality have its origin? Was there a teratogenic exposure during a critical period of organogenesis? Has a new mutation occurred in one of the parents’ germ cells? Are some of the DNA alterations in this newborn ancient relics that only now reveal themselves in the living world? Is this congenital anomaly a clue to the presence of other pathology? Will our best efforts have a reasonable chance of ensuring long-term health? Will siblings or offspring be similarly affected
Over the past several decades, research has dramatically improved our insights into the genetic and environmental causes of many isolated birth defects, multiple congenital anomaly syndromes, and other genetic conditions. In some instances, the molecular pathology has been dissected in impressive detail, such as for cystic fibrosis (CF) and the CF transmembrane regulator gene on chromosome 7q31. For other conditions, such as the VACTERL association, we have yet to understand the genomic origins but have accumulated much clinical and epidemiologic data that allow us to formulate helpful diagnostic criteria, predict prognosis, and estimate recurrence risks. For any individual newborn with a congenital anomaly, establishing an accurate diagnosis is the key to intelligent clinical management in both the short and long term.
In a busy nursery, caring for a newborn with a congenital malformation is nearly a daily activity. Although the consultative services of a clinical geneticist or dysmorphologist often are quite helpful, this resource may not be available, and the attending pediatrician or neonatologist must assume that role, at least temporarily. Naturally, such a task brings many challenges—cognitive, managerial, and emotional—for which the practitioner often feels underprepared. Useful tools include a careful prenatal history, three-generation pedigree, meticulous physical examination, and sensitivity for the emotional and social impact of the birth of a malformed child. A dramatically visible departure from the norm multiplies a parent’s fear, concern, worry, and guilt and might also become very stressful for nurses and physicians. Consequently, to optimize care for the infant and to instill confidence and control in an intrinsically unsettling situation, the clinical leaders of the health care team should prepare themselves by becoming familiar with a general strategy for diagnosis and management of an infant with congenital malformations.
In aggregate, birth defects are ubiquitously common and have likely been so throughout human history. Epi-demiologic studies consistently place the incidence of major malformations in newborns at 2% to 3% (1,2). Many other neonates harbor occult anomalies that are eventually detected later in childhood, giving a cumulative rate of major birth defects of approximately 4%, or one of every 25 newborn children. In the United States, 150,000 children with birth defects are born each year. Congenital anomalies are the leading cause of infant mortality and the second leading cause of death in children between the ages of 1 and 4 years (3). Excluding the intangible costs of pain and suffering, the lifetime economic cost per child in 1992 ranged from $75,000 to $503,000 (4). The Centers for Disease Control and the National Birth Defects Prevention Network conduct and facilitate a number of research and surveillance activities, central to which is the commitment of individual practitioners to diagnose and report all congenital defects accurately and reliably.
DEFINITIONS AND CLASSIFICATIONS
A congenital anomaly is any alteration, present at birth, of normal anatomic structure. It may be major or minor, isolated or part of a larger constellation of defects, of clear or uncertain cause. Several genetic and environmental etiologies are well delineated (Table 38-1), but the fundamental etiology of nearly half of all birth defects is unknown (5).
The nomenclature for various birth defects and malformation syndromes can be a source of confusion for both the novice and veteran clinician alike. Categories of anomalies
may seem arbitrary and capricious, but a developmental or embryologic perspective often will cast light on the rationale behind a particular designation. The term “birth defect,” enjoys wide usage and conveys immediate meaning for parents. “Congenital anomaly” is fundamentally equivalent, indicating an abnormality of anatomic structure present at birth and may be further refined in terms of severity (“major” and “minor”), pathogenesis (“malformation,” “deformation,” “disruption,” “dysplasia”), or pattern (“isolated,” “syndromic”). These terms are defined in Table 38-2.
may seem arbitrary and capricious, but a developmental or embryologic perspective often will cast light on the rationale behind a particular designation. The term “birth defect,” enjoys wide usage and conveys immediate meaning for parents. “Congenital anomaly” is fundamentally equivalent, indicating an abnormality of anatomic structure present at birth and may be further refined in terms of severity (“major” and “minor”), pathogenesis (“malformation,” “deformation,” “disruption,” “dysplasia”), or pattern (“isolated,” “syndromic”). These terms are defined in Table 38-2.
TABLE 38-1 CAUSES OF MALFORMATIONS IN NEWBORNSa | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
The great majority of congenital anomalies occur in isolation, as a single phenomenon, and are postulated to arise because of a primary, intrinsic malformation of a fetal structure that occurs at 10 weeks of gestation or earlier. On occasion, other family members are similarly affected, implying the presence of a single gene mutation that behaves as an autosomal dominant, autosomal recessive, or X-linked trait. More commonly, however, the family history is entirely bereft of other affected individuals. Classic mendelian genetics does not adequately explain this situation, but another model—multifactoral inheritance—has proven quite useful.
TABLE 38-2 Terminology | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
In the multifactoral model, a constellation of influences, including multiple genes inherited from each parent, and poorly defined environmental influences, seems to allow a developing fetal structure to cross a threshold of “liability,” beyond which morphogenesis proceeds abnormally (6). These birth defects tend to recur at a low rate, approximately 3% to 5% for each subsequent pregnancy for the parents of one affected child, 10% to 15% if two siblings have previously been similarly affected.
Multiple congenital malformations, on the other hand, are caused by a very different spectrum of fundamental problems. For a child with several major anomalies, the underlying cause is more likely to be a malformation sequence, developmental field defect, association, or syndrome, each of which may in turn may be caused by a chromosomal anomaly, a single-gene mutation, a teratogen, or unknown factors. Minor anomalies deserve special attention because, in terms of diagnostic significance, they are frequently the equals of the major malformations (7), often providing the linchpin of clinical recognition of a rare
syndrome. A single minor anomaly is present in 13% of neonates, but more than one is decidedly less common—two minor anomalies occur in only 1%, and three minor anomalies in just 0.05% (8). As the numbers of minor anomalies increase, the search for occult major anomalies and the consideration that a syndromic diagnosis is possible should increase proportionately.
syndrome. A single minor anomaly is present in 13% of neonates, but more than one is decidedly less common—two minor anomalies occur in only 1%, and three minor anomalies in just 0.05% (8). As the numbers of minor anomalies increase, the search for occult major anomalies and the consideration that a syndromic diagnosis is possible should increase proportionately.
MANAGEMENT STRATEGY
Since the last edition of this textbook, the American College of Medical Genetics (ACMG), under the sponsorship of the New York State Department of Health, has published clinical guidelines for health care practitioners who care for newborn infants with one or more birth defects (9). This document incorporates expert opinion from a broad array of disciplines and describes practical, detailed components of the history, physical examination, differential diagnosis, diagnostic stratagem, genetic counseling, and record keeping. As such, the ACMG guidelines represent a significant step forward in promoting a national standard of care with which neonatologists, pediatricians, and family practitioners should become familiar.
The fundamental approach to managing an infant with one or more congenital anomalies is much the same as the management of any other clinical scenario. Effective clinical intervention is organized around an understanding of the natural history of the condition at hand. History taking begins with conception and includes a detailed three-generation pedigree. Physical features must be scrutinized, measured, and documented with precision, and confirmatory studies must be carefully chosen and accurately interpreted. Common pitfalls include incomplete ascertainment of all relevant information, impetuous diagnosis and prognostication, and failure to communicate with parents in a straightforward and compassionate manner.
Neonates with congenital malformations present with extremely variable clinical needs, so a “one size fits all” strategy for management has limited practical value. However, a number of algorithmic approaches have proven useful. In the ACMG clinical management algorithm (Fig. 38-1), an early determination of whether the malformation is isolated or multiple helps organize subsequent clinical activity. Beyond this, the major thrust of activity is devoted to pursuing and establishing a precise diagnosis, the essential starting point for understanding natural history, designing effective intervention, and educating the family about recurrence risks and prenatal management options for subsequent pregnancies. Another, more integrated approach suggested by Dr. Judith Hall (10) delineates three parallel and simultaneous lines of activity, as outlined in Fig. 38-2. Urgent interventions for the infant are undertaken immediately. Also, from the outset, the family is provided with support in the form of information, preliminary interpretation, and acknowledgment of their distress and concern. The third concurrent activity is data collection—history, physical, laboratory studies and imaging, and preliminary definition of the nature of the problem. All efforts should be made to verify information and to amass a complete database.
History
Prenatal information, beginning with conception, must be specifically sought from both the mother and her obstetric records. The nature and timing of maternal illnesses, such as rubella or cytomegalovirus infections, including febrile episodes, can suggest a direct infectious disruption. Maternal use of alcohol, drugs, medications, or tobacco—degree, gestational timing, and duration—should be documented. Quickening, the date at which there is maternal sensation of fetal movement, and the subsequent vigor and frequency of fetal activity, reflects central nervous system integrity and peripheral muscular function. Prenatal ultrasonography, especially if performed serially and in detail, may yield critical information regarding the amount of amniotic fluid and malformations of the kidneys, brain, heart, skeleton, and gastrointestinal system. Recent reports have suggested an association between in vitro fertilization and several genetic conditions, such as Angelman syndrome (11), retinoblastoma (12) and Beckwith-Wiedemann syndrome (13). Chorionic villus sampling has previously been suspected to cause limb-reduction deformities, but recent studies by the World Health Organization affirm its safety for first-trimester prenatal diagnosis (14). A review of the ultrasound studies and the obstetric record, and a discussion with the attending obstetrician or perinatologist, can save valu-able time and effort. Maternal serum and amniotic fluid αfetoprotein levels and prenatal chromosome studies should be verified and documented in the infant’s medical record. Late trimester problems with fetal position, such as transverse or breech lie, potentially indicate neuromuscular or structural abnormalities of the fetus. Fetuses with significant neuromuscular problems often encounter perinatal distress and have a poor transition to extrauterine life. Finally, it often is useful to inquire about any prenatal event or factors that the parents suspect, even remotely, may have caused their infant’s problems. Concerns about witnessing a solar or lunar eclipse, for example, are best made explicit, if only for the purpose of assuaging guilt.
Family History
A systematic and detailed inquiry into the age, health, development, and congenital anomalies of all members of the immediate family comprises the core of an adequate family history. All second-degree relatives, such as grandparents, aunts, uncles, nieces, and nephews, should be considered, and more distant relatives may contribute valuable data. A three-generation pedigree provides a concise picture of patterns of inheritance. One must specifically clarify the biologic parentage for each individual. Spontaneous abortions, miscarriages, stillbirths, and infant deaths clearly are germane, but parents typically omit this information unless the interviewer specifically inquires. Consanguinity also should be directly but tactfully questioned. A “quick pedigree” is an
oxymoron; routinely the entire process requires patience and time. With this in mind, the interviewer will be sympathetic, on the day of delivery, to parents who are understandably terrified, exhausted, and disoriented. After rest, time, and a chance to speak with relatives, they may be more prepared to help refine and expand on their complete family history.
oxymoron; routinely the entire process requires patience and time. With this in mind, the interviewer will be sympathetic, on the day of delivery, to parents who are understandably terrified, exhausted, and disoriented. After rest, time, and a chance to speak with relatives, they may be more prepared to help refine and expand on their complete family history.
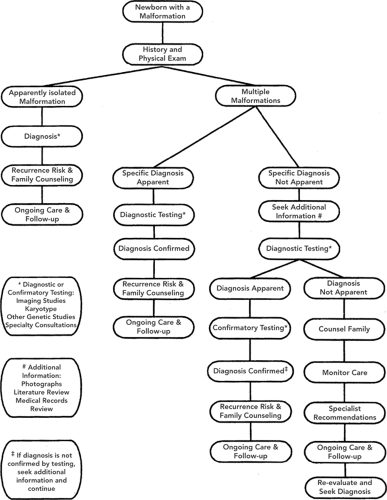 Figure 38-1 Management algorithm for the infant with multiple congenital anomalies. Reprinted from American College of Medical Genetics Foundation, sponsored by the New York State Department of Health. Evaluation of the newborn with single or multiple congenital anomalies: a clinical guideline. May 1999. Available at http://www.health.state.ny.us/nysdoh/dpprd/main.htm. Accessed 1/1/05, with permission. |
Physical Examination
It is axiomatic that a proper newborn physical examination is detailed and complete. The careful observer, especially one who repeats the examination several times, will recognize departures from the norm. Several points are worth keeping in mind when examining infants with a malformation or a generalized dysmorphic appearance.
Be alert. There is a heightened chance that additional, initially unsuspected anomalies are present. Although a malformation may be minor in severity, it might represent the most important, critical clue to the diagnosis
Collect “clues” systematically, by closely examining each topographical segment of the body and by scrutinizing progressively more detailed regions. For example, on
initial observation it may be noted that the fingers are disproportionately short; a closer examination may reveal that the fourth and fifth fingers are stiff and rigidly extended, with faint flexion creases, especially at the distal interphalangeal joints, that the nails are short and narrow, almost absent on the fifth finger. The placenta may be examined with the obstetrician or pathologist to seek evidence of cryptic twinning, umbilical cord anomalies, or amniotic bands
Document the examination with great care, using appropriate morphologic terms and sufficient detail, and strongly consider supplementing written findings with clinical photographs. Parental consent for photographs should be documented.
Measure those features that are obviously or potentially abnormal in size, shape, position, or symmetry. Normal standards are available for virtually any anatomic structure, encompassing all ages from preterm infant to adulthood (15,16). Hall (7) admonishes, “Never make a clinical judgment on a measurable parameter without measuring it.”
Examine both parents, if possible, seeking any signs of similar anomalies. Dominant conditions frequently manifest a subtle but distinctive phenotype in adults.
 Figure 38-2 Integrated approach to management. Modified from reference 48. |
Adjunctive Investigations
The emerging clinical picture will dictate imaging studies and consultations with pediatric subspecialists. For example, a newborn with Down syndrome, even when a cardiac murmur is absent, merits the attention of a pediatric cardiologist, because significant structural defects of the heart are present in 50% but may be missed on clinical examination. A newborn girl with puffy hands and feet, a webbed neck, and coarctation of the aorta also should receive a renal ultrasound, because kidney malformations commonly are associated with Turner syndrome. When the diagnosis is unclear and several malformations are present, occult anomalies of the central nervous system, heart, kidneys, vertebrae, and eyes are reasonable to pursue, especially when the known birth defects are multiple and severe. Renal ultrasounds are often ordered when the neonate has a single umbilical artery or an ear malformation, such as a preauricular pit. In the absence of supporting findings, such as other malformations, a family history of deafness or renal anomalies, or maternal diabetes, this investigation is unlikely to be useful (17). Skeletal films, to include hands, feet, long bones, pelvis, vertebrae, chest, and cranium, are helpful when length is less then the 5th percentile for gestational age or when the limbs are disproportionately short. These studies often require interpretation by a pediatric radiologist skilled in this area.
Chromosomal Analysis
Forty-six chromosomes are present in most normal human cells. Formation of ova and sperm, however, is a surprisingly error-prone process: nearly two-thirds of all fertilizations result in aneuploidy or abnormal chromosomal number or structure, with subsequent reproductive loss. Some of this prenatal loss occurs late enough to be recognized as a miscarriage or spontaneous abortion, but most wastage is occult. Ninety-eight percent of these chromosomal defects are lethal (13).
An abnormal karyotype occurs in one of 170 liveborn infants. Among chromosomally abnormal neonates, one-third have an extra sex chromosome with mild or no phenotypic manifestations in the newborn period, one-fourth have trisomy of an autosome, such as trisomy 21 or trisomy 18, and 40% have a variation of chromosomal structure, such as a deleted or duplicated segment or a translocation. Of the latter, most (79%) are balanced and generally do not cause birth defects. Approximately 10% of infants who die in the perinatal period secondary to multiple congenital malformations have abnormal cytogenetic studies (13).
Which infants deserve chromosomal studies? Truly isolated malformations are very infrequently caused by a cytogenetic defect. On the other hand, neonates with multiple major malformations or a generalized dysmorphic appearance, and stillborn infants, with or without malformations, should have cytogenetic testing. Between these extremes lies a sizable gray area. Factors in favor of chromosomal testing include intrauterine growth retardation, an abnormal neurologic exam, a major malformation accompanied by several minor malformations, and a history
of multiple pregnancy losses for the mother or in close relatives.
of multiple pregnancy losses for the mother or in close relatives.
Peripheral blood lymphocytes are the tissue of choice for most cytogenetic analysis, but many other tissues can be used, including skin fibroblasts, bone marrow, placenta, and pericardium, usually harvested postmortem. Two to three milliliters of venous or arterial blood, collected in a sodium heparin (green top) tube, are generally sufficient and should be kept at room temperature or refrigerated, never frozen, in transit. The cells usually will remain viable for 1 or 2 days, but the shortest possible transit time increases the chances for useful results. If the sample is shipped, an overnight courier is recommended. The lymphocytes are separated, incubated in the presence of a mitogen to stimulate cell division, which is then abruptly halted with colchicine, and the cell membranes are disrupted gently while being placed on a glass slide. After enzymatic preparation and staining, the “spread” of chromosomes is analyzed in approximately 20 cells. Generally, 550 or more bands are visible with Giemsa staining of 46 chromosomes at metaphase. Several cells are photographed and arranged in standard groupings, a karyotype. Turnaround time for cytogenetic analysis is typically three to four days, although some laboratories are able to provide results in slightly less time. Because of the high percentage of dividing cells in bone marrow, karyotypes from this tissue may be obtained in a matter of hours, although the quality of the banding frequently is inadequate for high-resolution analysis of small or subtle abnormalities. To achieve the latter, a special request for prometaphase analysis should accompany a peripheral blood sample.
In the past several years, an increasing number of syndromic conditions have been found to be caused by very small chromosomal deletions that are not visible by routine karyotyping, even at pro-metaphase levels of detail. Molecular probes that will hybridize at these loci with great specificity have been developed. These DNA probes are complexed with a fluorescent marker and become powerful tools for detecting submicroscopic chromosomal deletions. This technique, termed fluorescent in situ hybridization (FISH), has also been adapted for whole, entire chromosomes and can identify the nature of many chromosomal anomalies, such as translocations. A metaphase spread can, in essence, be painted with several single-locus or whole-chromosome FISH probes simultaneously, providing a highly specific, and colorful, picture of genomic structure.
Among children with mental retardation of unknown etiology, in whom extensive investigations, including standard cytogenetic studies, are normal, approximately 5% harbor very small, occult chromosomal deletions or unbalanced translocations. A recently developed technique—subtelomeric probe analysis—uses FISH and/or other molecular tools to ascertain whether the regions just proximal to the tips of each of the 46 chromosomes are present in their normal locations. Although initially designed to investigate subnormal intelligence in older children, subtelomeric analysis appears to also be useful for some infants with congenital anomalies in whom standard investigations have not been fruitful. This technology seems to have better diagnostic success for individuals with intrauterine growth retardation, microcephaly, and a positive family history (19).
Metabolic Studies
Inborn errors of metabolism are often assumed to have a purely biochemical or neurologic phenotype, but metabolic disease is well recognized as an occasional cause of dysmorphic facial features and congenital malformations (20). For instance, ambiguous genitalia are seen in some cases of 21-hydroxylase deficiency and other types of congenital adrenal hyperplasia, and infants with pyruvate dehydrogenase deficiency may have agenesis of the corpus callosum and facial features that resemble fetal alcohol syndrome. A number of the congenital disorders of glycosylation feature congenital malformations of the heart, limbs, and central nervous system (21). Many peroxisomal conditions result in distinctive phenotypes: Zellweger syndrome, also known as cerebro-hepato-renal syndrome, and rhizomelic chondrodysplasia punctata (RCDP) exemplify this class of disease. For the Zellweger spectrum of peroxisomal abnormalities, serum levels of very long chain fatty acids will be elevated; in RCDP the very long chain fatty acids are normal but phytanic acid is elevated.
Synthesis and Analysis of Data
Every clinician develops a unique, individual strategy of diagnosis. Acquiring a deep and broad fund of knowledge is arguably fundamental and, oftentimes, sufficient in itself. Unfortunately, the sheer numbers of conditions and syndromes impose some limits on the “brute force” approach; the London Dysmorphology Database, for instance, contains over 3,000 multiple congenital anomaly syndromes (22).
Precise diagnosis is neither possible nor necessary in the immediate newborn period for many neonates with a multiple congenital anomaly syndrome. More than half of all individuals with congenital anomalies never receive a firm diagnosis, even after the most definitive of workups, and remain an “unknown.” Categorization of an anomaly as a malformation, disruption, or deformation, however, is a feasible and useful first step. These terms have been defined and discussed previously. An isolated major anomaly in the absence of any similarly affected relative would suggest a multifactoral etiology. If there are multiple malformations, one of the following strategies may be useful:
Instant recognition, or gestalt diagnosis, which depends on the clinician’s previous experience and strength of visual memory. Certain caveats apply, however: many disorders have a considerable range of phenotypic variation, and other conditions, or phenocopies, may mimic the one that has instantly come to mind
Perusal of an atlas or illustrated text, such as Smith’s Recognizable Patterns of Human Malformation, to match a photograph with the patient. This simple strategy often yields excellent results.
Pattern analysis, in which all phenotypic and clinical “problems” are enumerated, grouped, combined, recombined, and weighed to discern developmental relationships, sequences, and influences. Major organ systems or classes of disease (e.g., skeletal dysplasias) then become entry points for further comparison, matching the patient’s pattern against published descriptions while attempting to take into account phenotypic variability
Focusing the initial investigation on the anomaly that is most distinctive, rare, or unusual. Clinodactyly of the fifth finger is very common, but a coloboma of the iris is fairly unusual. A variety of texts or electronic databases then can be consulted and a relatively short list of diagnostic possibilities generated.
Once a preliminary analysis has generated a differential diagnosis, all reasonable efforts are made to test each competing hypothesis. Often, a clinical finding can corroborate a possibility. For instance, a lateral radiograph of the knee may allow confirmation of chondrodysplasia punctata by demonstrating the typical stippled, punctate mineralization of the epiphyses. Although many diagnoses are purely clinical, molecular tools are becoming extremely helpful. An electronic literature search or querying an internet resource, such as Online Mendelian Inheritance in Man, can help determine whether a novel approach using direct deoxyribonucleic acid (DNA) sequence analysis, FISH, or linkage analysis has been developed for the disease in question.
More often than not, the diagnostician will not be able to establish an etiology. When this is the case, there is a temptation to “force” a diagnosis, analogous to hammering a somewhat square peg into a slightly round hole. This may not be in the best interests of the patient so that, in these situations, there is honor in admitting ignorance. Certain characteristics of many syndromic conditions, such as the so-called “elfin” facies of Williams syndrome, are not apparent in the newborn period. The most useful diagnostic decision may be to wait and start afresh at a later time, recollecting data, recombining features into new patterns, and researching the literature. The assistance of a clinical geneticist or dysmorphologist during all phases of evaluation frequently optimizes the diagnostic process. If such services are not immediately available via direct consultation, telephonic or telemedicine consultation may be highly effective.
Other Management Issues
Communication with the parents of a child with congenital anomalies requires a compassionate, timely, and honest presentation of the facts. One’s choice of terminology is important but often challenging. Many parents find it quite helpful for the principal caregiver to examine the infant in their presence, pointing out the features that seem to be unusual, and delineating those that are normal. Parents naturally feel responsible for the birth defect, which they might interpret as a reflection of their own shortcomings, real or imagined. Guilt is as common for the parents of a malformed child as pride is for the parents of a normal newborn. Although it is not always possible to convince parents to set aside unreasonable guilt, they can at least be reassured that they had no control over the events causing the abnormality and that they have permission to not feel guilty.
Expect the full spectrum of grief from both parents. They have experienced the loss of a much-anticipated “normal” child. Shock, denial, bargaining, and acceptance will all occur, even when their child does not have a lethal condition. The physicians and nursing personnel involved in the care of the malformed infant work as a team to keep track of this process and be alert for dysfunctional grief. Social work services, clergy, and support groups are important adjuncts. The National Organization for Rare Disorders, the Alliance of Genetic Support Groups, and specific support groups can provide up-to-date information and lay contacts for interested parents.
Not uncommonly, parents and relatives seek and find extensive information about birth defects, syndromes, chromosomal anomalies, and teratogenic conditions on the internet. In some cases, this information is more detailed and current than that available to a busy clinician who relies on brief textbook entries. Based on this information, parents may become firm, even strident and argumentative, demanding specific tests and management. This situation requires judgment, tact, and an open mind on the part of the attending physician, because this information may be quite useful, and parents who remain your allies will positively influence the entire spectrum of medical management. As correct and cutting edge as this information may be, parents will appreciate your pointing out that the clinical acumen and experience of the neonatal team is essential to assess the extent to which generic recommendations may be helpful for any particular patient. An imperative component of this process will be to verify the reliability of the information and ascertain whether it is based on credible scientific data.
If the malformed infant dies and there is any question regarding the precise diagnosis, a full, unrestricted autopsy can prove extremely useful. Visceral, central nervous system, and skeletal anomalies frequently come to light at postmortem examination. Clinical photography and cytogenetic analysis of fibroblasts obtained from sterile skin biopsy, fascia, or pericardium also may yield important insights. Tissue, cells in culture, and extracted DNA can be stored in a long-term repository and later reanalyzed in light of new research or collaboration.
Families often are reluctant to grant permission for an autopsy. In many instances, however, this procedure has profound implications for the parents’ reproductive options and even those of distant relatives. Cultural and social beliefs and practices must, of course, be carefully respected regarding the care of the child’s body after death. Nevertheless, an autopsy can be recast in the light of a final gift of the child to his family, perhaps even to the world, if in fact a diagnosis is thereby established or medical science advanced.
SELECTED EXAMPLES
Teratogenic Conditions
A teratogen, from the Greek root teras, meaning monster or marvel, is any environmental factor that causes a structural or functional abnormality in the developing fetus or embryo. These environmental agents include infections, medications, drugs, chemicals, and maternal metabolites, such as phenylalanine (Table 38-3). By their very nature, teratogens induce a disruption or sequence of disruptions of inherently normal tissue. Extensive compendia of these agents have been studied in humans and laboratory animals, and several excellent resources are available for the clinician. Additionally, both professionals and patients can access regional teratogen hot lines.
Ethanol, the most common human teratogen, is estimated to affect as many as 1 in 300 newborns, primarily as a neurotoxin, with consequences ranging from cerebral palsy to learning disability. Up to one-fifth of mental retardation (usually mild) is attributable to fetal alcohol syndrome (FAS), but numerous structural anomalies have also been reported (Table 38-4). Although the syndrome has been recognized for over 30 years, the dysmorphisms and other clinical signs of FAS are still often overlooked in the newborn nursery (23).
Multifactoral Disorders
The multifactoral model of inheritance, as previously discussed, provides a conceptual basis for understanding the pathogenesis and recurrence risks of isolated, nonsyndromic congenital malformations. The central concept of this model is that multiple genes and environmental factors influence whether a particular anatomic structure may develop abnormally. The susceptibility, or genetic liability, of a malformation in a population is described in terms of
a continuous distribution of susceptibility factors in which there is a point, or threshold, beyond which, in an all-or-none fashion, a structural defect will occur. Table 38-5 lists some common multifactoral disorders and their recurrence risks. This type of multifactoral heritability is postulated to account for most isolated malformations.
a continuous distribution of susceptibility factors in which there is a point, or threshold, beyond which, in an all-or-none fashion, a structural defect will occur. Table 38-5 lists some common multifactoral disorders and their recurrence risks. This type of multifactoral heritability is postulated to account for most isolated malformations.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


