Coagulopathies and Sickle Cell Disease
Biochemistry and Physiology of Hemostasis
The stimulus that initially causes clot formation occurs as a consequence of disruption of endothelial cells. This leads to exposure of collagen and subendothelial tissues. The hemostatic response to tissue injury consists of four stages. First, vasoconstriction by the contraction of smooth muscle in the injured vessel wall reduces blood flow. Second, platelets adhere to the exposed endothelium, aggregate, and release their granular contents. This activity stimulates further vasoconstriction and recruits more platelets. This action results in ‘primary hemostasis’ that occludes the gap in the blood vessel and stops blood loss through the vessel. Third, the extrinsic and intrinsic coagulation systems are activated to form fibrin, which stabilizes the platelets and prevents disaggregation. Fourth, fibrinolysis results from the release of plasminogen activators from the injured vessel wall. These activators limit the coagulation process. Once healing has taken place, they begin dissolution of formed clot so that vascular patency can be restored.1
Endothelial Cells
Endothelial cells maintain the integrity of the blood vessel and prevent extravasation of blood into the surrounding tissue.1 Passive thromboresistance is provided by endothelial proteoglycans, primarily endogenous heparin sulfate. Active thromboresistance is achieved through several mechanisms, including the synthesis and release of prostacyclin, a potent vasodilator and an inhibitor of platelet adhesion and aggregation.1,2
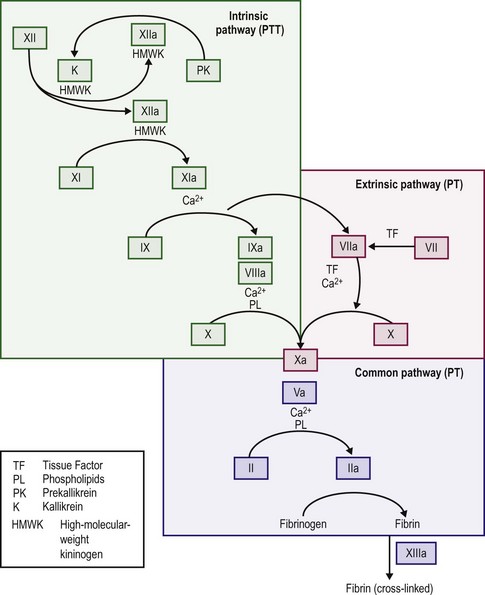
When the endothelium is injured, tissue factor (thromboplastin) is produced and rapidly promotes local thrombin formation.3 Tissue factor binds factor VII and converts it to factor VIIa (Fig. 5-1), which is the first step in activation of the extrinsic coagulation pathway. It also activates factor IX, which is the major activator of the common pathway, resulting in fibrin formation.4 Capillaries seal with little dependence on the hemostatic system, but arterioles and venules require the presence of platelets to form an occluding plug. In arteries and veins, hemostasis depends on both vascular contraction and clot formation around an occluding primary hemostatic plug.5
Platelets
In the resting state, platelets circulate as disk-shaped, anuclear cells that have been released from megakaryocytes in the bone marrow. They are 2–3 µm in size and remain in circulation for approximately five to nine days unless they participate in coagulation reactions or are removed by the spleen. Normal blood contains 150,000–400,000 platelets/µL.5 In the resting state platelets do not bind to intact endothelium.
Platelet Adhesion
Once platelets bind to injured tissue and are activated, their discoid shape changes. They spread on the subendothelial connective tissue and degranulate releasing serotonin, adenosine diphosphate, adenosine triphosphate, and calcium, and alpha granules release factor V, fibrinogen, von Willebrand factor (FVIII : vWF), fibronectin, platelet factor 4, β-thromboglobulin, and platelet-derived growth factor.6,7 This recruits and aggregates more platelets from the circulation onto the already adherent platelets.7
When a vessel is disrupted, platelet adhesion occurs through the binding of collagen and vWF (found in the subendothelium) to the platelet membrane (Fig. 5-2). For platelet adhesion to occur, platelets must express specific glycoprotein Ib receptors on their surface to bind the vWF complex. If this specific glycoprotein is missing, platelets are unable to adhere to areas of injury.8 Platelets in Bernard–Soulier syndrome lack glycoprotein Ib and are unable to adhere and form the initial hemostatic plug.9 If the vWF is defective or deficient, platelets do not adhere to sites of vascular injury. The result is von Willebrand disease, of which several specific types and subtypes have been defined.10–12 Very high concentrations of prostacyclin also can inhibit platelet adhesion to exposed subendothelium.5
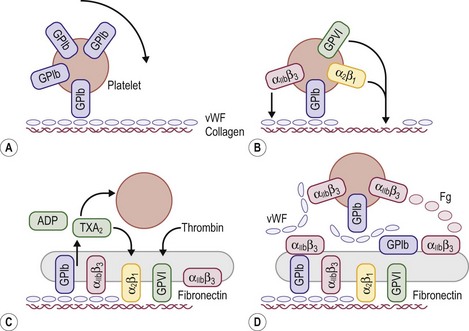
FIGURE 5-2 Schematic representation of platelet adhesion and aggregation under flow conditions. (A) Rolling of platelets over collagen-bound vWF mediated by GPIb. (B) Firm attachment mediated by α2β1 and glycoprotein VI (GP VI) binding to collagen, and by αIIbβ3) binding tocollagen-bound vWF. (C) Platelet activation, secretion, and spreading. (D) Aggregate formation.
Platelet Aggregation
Aggregation is a complex reaction that involves platelet granule release, cleavage of membrane phospholipids by phospholipases A2 and C, alterations in intracellular cyclic adenosine monophosphate levels, mobilization of intracellular calcium, and the expression of fibrinogen receptors on the platelet surface. If fibrinogen receptors (glycoproteins IIb and IIIa) or fibrinogen are missing, platelets do not aggregate.13,14 This results in Glanzmann thrombasthenia causing patients to have a serious, life-long bleeding disorder.6
After aggregation, platelets function to enhance thrombin formation. The platelet membrane provides specific binding sites for factors Xa and V causing effective assembly of the prothrombinase complex making thrombin.7 Thrombin formation makes a stable hemostatic plug of adherent platelets surrounded by a network of fibrin strands.
Generation of Thrombin
The majority of thrombin production results from the activation of the intrinsic coagulation system, not the extrinsic system. Exposed subendothelium converts factor XII to factor XIIa and thereby activates the intrinsic pathway, although deficits in factor XII do not cause a bleeding disorder. Activation of factors XI and IX follows, and activated factor IX in combination with factor VIII, calcium, and platelet phospholipid activates factor X. Activated factor VII, complexed with tissue factor, activates factor IX. Factor Xa with factor V then cleaves prothrombin into the active molecule thrombin, which can convert fibrinogen into fibrin.4,15
Formation of Fibrin
When thrombin acts on fibrinogen, fibrin monomers result after the proteolytic release of fibrinopeptides A and B. The monomeric fibrin then polymerizes into a gel.4,15 With additional stabilization of the fibrin gel provided by factor XIII, fibrin surrounds and stabilizes the platelet plug. This process makes the multimeric fibrin more resistant to plasmin digestion and completes the formation and stabilization of the blood clot.16
Several regulatory proteins serve to localize thrombin formation to the surface of the blood vessel. Endothelial cells have receptors for protein C. Protein S is a co-factor for the activation of protein C. Thrombomodulin is an endothelial surface protein that acts in combination with thrombin to activate the bound protein C. Activated protein C then degrades factors Va and VIIIa, which inhibit thrombin formation.17
Heparin-like anticoagulant molecules, present on endothelial cells, act in combination with antithrombin III to inhibit factors XIIa, XIa, IXa, and Xa and thrombin. Inhibition of these factors prevents the spread of clot to uninjured adjacent vessels and the blockage of large vessels by excessive clot formation.15,17 Endothelial cells, as mentioned previously, produce PGI2 (prostacyclin), a potent vasodilator and inhibitor of platelet aggregation and adhesion.
Fibrinolysis
The regulatory process that dissolves fibrin and preserves vessel patency is called fibrinolysis. Circulating plasminogen is converted into plasmin by tissue plasminogen activators. These activators are released from the vessel walls at the site of blood clotting. They bind to the fibrin clot and convert plasminogen to plasmin. Plasmin enzymatically degrades fibrin, fibrinogen, and other plasma proteins, and this process results in the dissolution of formed clot.15,17
Clinical Evaluation
There is currently no completely reliable screening test available to evaluate hemostasis in the preoperative patients. A careful history, including a full family history, remains the best means of uncovering mild bleeding problems, including von Willebrand disease or qualitative platelet abnormalities.18 These disorders may easily escape standard laboratory screening procedures, such as prothrombin time (PT), activated partial thromboplastin time (aPTT), platelet count, and bleeding time. aPTT screening yields many false-positive results caused by both analytical problems and detection of clinically insignificant disorders. In addition, a normal aPTT may lead to a false sense of safety because it does not exclude all serious bleeding disorders. Because no method can reliably predict all bleeding complications, postoperative monitoring remains important for all patients.19 Likewise, patients with mild disorders who have not previously undergone an operation may have no history of bleeding problems and might be identified preoperatively only if screening tests are performed.18 It is important to consider the history as the most important component of a diagnostic strategy and to investigate thoroughly any story of unusual bleeding, even if the screening tests are normal.20 Conversely, studies examining the utility of a screening preoperative PT and aPTT in patients undergoing tonsillectomy and adenoidectomy concluded that routine screening with a PT and aPTT for all patients regardless of history cannot be recommended.19,21 In obtaining a history from the patient and parents, positive answers to the questions posed in Box 5-1 indicate the need for further evaluation.18,22–24
If there is a history of abnormal bleeding, the following points must be established. The type of bleeding (i.e., petechiae, purpura, ecchymosis, and single or generalized bleeding sites) can give an indication of the underlying defect. Petechiae and purpura are most frequently associated with platelet abnormalities, either of function or numbers. Von Willebrand disease is most frequently associated with mucosal bleeding, including epistaxis, whereas hemophilia is most often associated with bleeding into joints or soft tissue ecchymosis, or both. Bleeding when the umbilical cord separates is most often associated with a deficiency in factor XIII, as is unexplained bleeding of the central nervous system.16,25 A single bleeding site, such as repetitive epistaxis from the same nostril, is frequently indicative of a localized, anatomic problem and not a system-wide coagulation defect.
Any previous or current drug therapy must be fully documented, and a search is made for over-the-counter medications that the patient might be taking but does not consider ‘medicine’ and has therefore not mentioned. Aspirin, ibuprofen, cough medications containing guaifenesin, and antihistamines can lead to platelet dysfunction or uncover a previously undiagnosed bleeding disorder such as von Willebrand disease.26,27 The presence of other medical problems including renal failure with uremia, hepatic failure, malignancies, gastrointestinal malabsorption, vascular malformations, cardiac anomalies with or without repair, and autoimmune disorders is essential to elicit because these may have associated coagulopathies.
Laboratory Evaluation
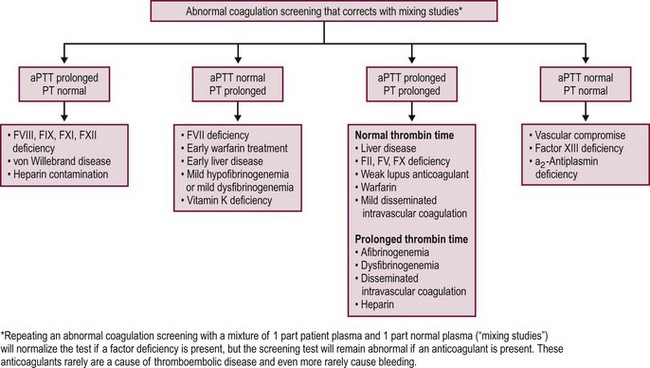
When the bleeding history and/or family history suggest the possibility of a bleeding disorder, or if it is impossible to obtain a history due to family or social circumstances, it is customary to proceed with a series of laboratory investigations to look for a possible bleeding diagnosis. Generally, screening tests are performed first and should include a blood cell count, PT, and aPTT (Fig. 5-3).20 Additional tests can measure fibrinogen levels, assess the thrombin time, screen for inhibitors of specific coagulation factors, measure specific factor levels, and test for platelet function and von Willebrand disease.20,28 Patients also can be evaluated for evidence of DIC by using multiple assays to test for the presence of various fibrinopeptides and products from the breakdown of fibrin or fibrinogen.
Platelet Count
The platelet count measures the adequacy of platelet numbers to provide initial hemostasis. Thrombocytopenia (a platelet count of <150,000/µL) is one of the most common problems that occur in hospitalized patients. As stated previously, typical manifestations include mucocutaneous bleeding. The risk of bleeding is inversely proportional to the platelet count. When the platelet count is <50,000/µL, minor bleeding occurs easily and the risk of major bleeding increases. Counts between 20,000–50,000/µL predispose to bleeding with even minor trauma; with counts <20,000/µL, spontaneous bleeding may occur; with counts <5000/µL, severe spontaneous bleeding is more likely. However, patients with counts <10,000/µL may be asymptomatic for years.29 Surgical bleeding does not usually occur until the platelet count is <50,000 platelets/µL.30 A platelet count of <50,000/µL is considered a cut-off criterion for transfusions, and the prophylactic use of platelet transfusion is indicated for any invasive procedure. Patients with significant clinical bleeding should also be transfused with platelets.30
Bleeding Time and the PFA-100 Analyzer
The bleeding time is defined as the length of time required for a standardized incision to stop oozing blood that can be absorbed onto filter paper. A variety of procedures have been used, but all have variable sensitivity and have been difficult to reproduce accurately, leading many centers to drop the bleeding time from the list of approved laboratory tests.31 The PFA-100 Analyzer (Siemans Healthcare Diagnostics, Deerfield, IL) is now widely used as a replacement for the bleeding time. It creates an in vitro high shear stress condition that results in the activation of platelet-dependent and vWF-dependent attachment and aggregation of platelets to a collagen–ADP or collagen–epinephrine surface. In most cases, the PFA-100 closure time is superior to the bleeding time in the detection of von Willebrand disease, aspirin effect, or platelet dysfunction.31 However, test results can be influenced by the sample’s hematocrit. Although the PFA-100 does not detect all platelet dysfunctions or cases of von Willebrand disease, when used in conjunction with a standardized questionnaire, it will likely detect impaired hemostasis in most cases.31,32
Prothrombin Time
The PT is a measure of the function of the extrinsic and common coagulation pathways. It represents the time (in seconds) for the patient’s plasma to clot after the addition of calcium and thromboplastin (an activator of the extrinsic pathway).33,34 Isolated prolongation of the PT is seen most commonly in patients who are deficient in vitamin K due to previous antibiotic treatment. It also occurs with factor VII deficiency, mild hypofibrinogenemia, dysfibrinogenemia, and warfarin therapy. The PT may also be prolonged with significant liver dysfunction.33,34
Partial Thromboplastin Time
The aPTT measures the function of the intrinsic and common coagulation pathways. The aPTT represents the time (in seconds) for the patient’s plasma to clot after the addition of phospholipid, calcium and an intrinsic pathway activator. The aPTT detects deficiencies in factors XII, XI, IX, and VIII and in the common pathway, but mild factor deficiencies may be missed. The aPTT also is used to monitor anticoagulation with heparin.33,34
Several inherited disorders of coagulation are not detected by the preceding tests. Results from standard hemostatic screening tests, such as the aPTT and international normalized ratio (INR) assessments, are normal in factor XIII (FXIII) deficiency. Therefore, assessment of clot stability is the most common screening test used for FXIII deficiency with a quantitative assay required to confirm the diagnosis of FXIII deficiency.35 Von Willebrand disease patients may have normal or prolonged aPTTs, and patients with a deficiency in α2-antiplasmin have a normal aPTT. Both the PT and aPTT are prolonged in patients with deficiencies of factors X and V, prothrombin, and fibrinogen and in patients with DIC or severe liver disease.34,36
Fibrinogen
The standard method for fibrinogen determination measures clottable fibrinogen by using a kinetic assay. Normal levels of fibrinogen are 150–350 mg/dL. As fibrinogen is the substrate for the final reaction in the formation of a clot and all plasma-based screening tests depend on the formation of a clot as the end point of the reaction, fibrinogen levels below 80 mg/dL prolong the PT, aPTT, and thrombin time and therefore make the results uninterpretable. Large amounts of fibrin degradation products interfere with the formation of fibrin and cause an artificially low level of measured fibrinogen. An immunologic-based assay for fibrinogen is used to measure both clottable and nonclottable fibrinogen. This test is most often used in identifying patients with a dysfibrinogenemia in whom the functional level of fibrinogen is low and the immunologic level is normal.34,36
Inhibitor Screening Tests
Repeating the PT or aPTT by using a 1 : 1 mix of patient plasma with normal plasma is a useful procedure for investigating a prolonged PT or aPTT. Normal plasma has, by definition, 100% levels of all factors. When mixed with an equal volume of patient plasma, if there is a minimum of 50% of any given factor present, the PT or aPTT should normalize. Correction of the clotting time suggests the presence of a factor deficiency whereas lack of normalization suggests the presence of an inhibitor that interferes with either thrombin or fibrin formation.33,34
Two types of acquired inhibitors prolong the aPTT. One blocks or inactivates one of the intrinsic factors, whereas the other is a lupus-like inhibitor that interferes with phospholipid-based clotting reactions. The first type of inhibitor occurs in 10–15% of hemophilia A patients and can occur spontaneously, but it is extremely rare in nonhemophiliac children.37 The lupus-like inhibitor is associated not with bleeding problems but rather with an increased risk of thrombotic problems in adults. Lupus-like inhibitors are mentioned because they commonly cause prolongations of the aPTT.38 Specific investigation of either of these situations should be referred to a coagulation reference laboratory.
Platelet Function Studies
Platelet function studies measure in vitro platelet aggregation. In this procedure, platelet-rich plasma is incubated with an agonist and then changes are noted in the amount of light transmitted through the platelet suspension. Agonists used to induce platelet aggregation include collagen, epinephrine, ADP, thrombin, and ristocetin. Three distinct phases are seen in the reaction. The first is an initial change in the shape of the platelets, leading to a temporary decrease in light transmission. Next is the first wave of aggregation, which is a reversible platelet-platelet interaction. With additional stimulation, the final phase—the second wave of aggregation—occurs and produces irreversible platelet aggregation. The second wave of aggregation is due to release of the platelet granules and thromboxane A2 synthesis. The release reaction is blocked by aspirin and is absent in patients with an inherited storage pool defect, congenital deficiency in thromboxane A2 synthesis, or cyclooxygenase deficiency.7 The PFA-100 has become the test of choice to replace the bleeding time and is used to screen for a variety of disorders, but full characterization of platelet function requires traditional platelet aggregation studies in a specialized laboratory.
Specific Factor Assays
Specific factor assays are available for all known coagulation, fibrinolysis, and anticoagulation factors to quantify their levels in plasma. These tests are not indicated unless a screening test result is abnormal. The only exception involves the patient with a history that is suggestive of von Willebrand disease, factor XIII deficiency, or dysfibrinogenemia. In these cases, the aPTT may not be sensitive enough to detect the disorder. Further testing may be justified on clinical suspicion based on the patient’s history.33,34 For von Willebrand disease, the workup consists of measuring factor VIII levels, vWF antigen levels, ristocetin co-factor activity, and ristocetin-induced platelet aggregation. Analysis of the distribution of vWF multimers can be useful to the hematologist in identifying the specific type of von Willebrand disease.10–12
Tests for Disseminated Intravascular Coagulation
The tests that are available in most hospital laboratories for identification of DIC are semiquantitative fibrin or fibrinogen degradation product assays, which involve a slide agglutination procedure or a D-dimer assay. An increased amount of these degradation products suggests that either plasmin has circulated to lyse fibrin and fibrinogen or the patient’s hepatic function is insufficient to clear the small amounts of regularly produced degradation products. The D-dimer test is a slide agglutination procedure that tests for the presence of two D subunits of fibrin that are cross-linked by factor XIII. This test provides specific evidence that plasmin has digested the fibrin clot and not fibrinogen. It is positive in patients with DIC, in patients with resolving large intravascular clots, and in patients with hepatic insufficiency. Specific assays to demonstrate the presence of soluble fibrin monomer complexes or fibrinopeptides produced by the conversion of prothrombin to thrombin are also useful in some situations and available in specialized laboratories.34,39
Hemophilia A and B
Hemophilia A and B are X-linked recessive bleeding disorders caused by decreased levels of functional procoagulant factor VIII (FVIII) and factor IX (FIX), respectively. Approximately 80% of all hemophilia patients have FVIII deficiency, which is classic hemophilia. The remaining 20% have FIX deficiency, which is called Christmas disease. These are rare disorders, with a prevalence of only 13.4/100,000 males.41 Until 1964, the treatment of hemophilia was limited by volume restrictions imposed by the use of whole blood or fresh frozen plasma. At that time, the FVIII-rich fraction of fresh frozen plasma (known as cryoprecipitate) was discovered.42 Specific lyophilized FVIII concentrates have since been developed, as have prothrombin concentrates containing factors II, VII, IX, and X. Concentrates containing only FIX for the treatment of hemophilia B patients and factor VIII/vWF concentrates for the treatment of von Willebrand disease have also been developed.43,44
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


