Chapter 412 Chronic Severe Respiratory Insufficiency
412.1 Neuromuscular Diseases
Neuromuscular diseases (NMDs) of childhood include muscular dystrophies, metabolic and congenital myopathies, anterior horn cell disorders, peripheral neuropathies, and diseases that affect the neuromuscular junction. Decreases in muscle strength and endurance resulting from neuromuscular disorders can affect any skeletal muscle, including muscles involved in respiratory function. Of particular concern are those muscles mediating upper airway patency, generation of cough, and lung inflation. Acute respiratory insufficiency is often the most prominent clinical manifestation of several acute neuromuscular disorders, such as high-level spinal cord injury, poliomyelitis, Guillain-Barré syndrome (Chapter 608), and botulism (Chapter 202). Although much more insidious in its clinical course, respiratory dysfunction constitutes the leading cause of morbidity and mortality in progressive neuromuscular disorders (e.g., Duchenne muscular dystrophy [Chapter 601], spinal muscular atrophy, congenital myotonic dystrophy, myasthenia gravis [Chapter 604], and Charcot-Marie-Tooth disease [Chapter 605]).
Treatment
Even though gene-targeted therapies are being developed for some NMDs, current interventions are primarily supportive rather than curative. Close surveillance through periodic review of the history and physical examination is critical. The development of personality changes, such as irritability, decreased attention span, fatigue, and somnolence, may point to the presence of sleep-associated gas exchange abnormalities and sleep fragmentation. Changes in speech and voice characteristics, nasal flaring, and the use of other accessory muscles during quiet breathing at rest may provide sensitive indicators of progressive muscle dysfunction and respiratory compromise. Although the frequency of periodic re-evaluation needs to be tailored to the individual patient, tentative guidelines were developed for patients with DMD; an abbreviated summary of such recommendations, applicable to all children with NMDs, is provided in Table 412-1.
Table 412-1 PROPOSED GUIDELINES FOR INITIAL EVALUATION AND FOLLOW-UP OF PATIENTS WITH NEUROMUSCULAR DISEASE
| INITIAL EVALUATION | BASIC INTERVENTION/TRAINING |
|---|---|
| History/physical/anthropometrics | Nutritional consultation and guidance |
| Lung function and maximal respiratory pressures (PFTs) | Regular chest physiotherapy |
| Arterial blood gases | Use of percussive devices |
| Polysomnography* | Respiratory muscle training |
| Exercise testing (in selected cases) | Annual influenza vaccine |
| If vital capacity >60% predicted or maximal respiratory pressures >60 cm H2O | Evaluate PFTs every 6 mo |
| CXR and polysomnography every year | |
| If vital capacity <60% predicted or maximal respiratory pressures <60 cm H2O | Evaluate PFTs every 3-4 mo |
| CXR, MIP/MEP every 6 mo | |
| Polysomnography every 6 mo to every year |
CXR, chest x-ray; MEP, maximal expiratory pressure; MIP, maximal inspiratory pressure; PFT, pulmonary function test.
* Please note that if polysomnography is not readily available, multichannel recordings including oronasal airflow, nocturnal oximetry, and end-tidal carbon dioxide levels may provide an adequate alternative.
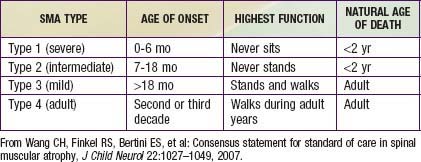
Guidelines for evaluation and management of patients with SMA were developed on the basis of expert consensus. Four classifications of SMA, based on age of onset and level of function, are recognized (Table 412-2). Treatment of SMA is focused on level of function (non-sitter, sitter, or walker) rather than SMA type. Unlike patients with DMD, patients with SMA do not demonstrate correlation between pulmonary function and need for mechanical ventilatory support. Rather, longitudinal monitoring for signs and symptoms of sleep-disordered breathing and ineffective airway clearance should be utilized to direct patient care.
Annane D, Orlikowski D, Chevret S, et al. Nocturnal mechanical ventilation for chronic hypoventilation in patients with neuromuscular and chest wall disorders. Cochrane Database Syst Rev. 2007;4:1-34.
Kennedy JD, Martin AJ. Chronic respiratory failure and neuromuscular disease. Pediatr Clin North Am. 2009;56:261-273.
Simonds AK. Recent advances in respiratory care for neuromuscular disease. Chest. 2006;130:1879-1886.
Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22:1027-1049.
412.2 Congenital Central Hypoventilation Syndrome
Genetics
The majority of CCHS cases occur because of a de novo PHOX2B mutation, but 5-10% of children with CCHS inherit the mutation from an asymptomatic parent who is mosaic for the PHOX2B mutation. CCHS is inherited in an autosomal dominant manner. Therefore, an individual with CCHS has a 50% chance of transmitting the mutation, and resulting disease phenotype, to each child. Mosaic parents have up to a 50% chance of transmitting the PHOX2B mutation to each offspring. Genetic counseling is essential for family planning and for preparedness in the delivery room for an anticipated CCHS birth. PHOX2B testing is advised for both parents of a child with CCHS to anticipate risk of recurrence in subsequent pregnancies. Further, prenatal testing for PHOX2B mutation is clinically available (www.genetests.org) for families with a known PHOX2B mutation.



