Chapter 348 Cholestasis
348.1 Neonatal Cholestasis
H. Hesham A-kader and William F. Balistreri
Neonatal cholestasis is defined biochemically as prolonged elevation of the serum levels of conjugated bilirubin beyond the 1st 14 days of life. Jaundice that appears after 2 wk of age, progresses after this time, or does not resolve at this time should be evaluated and a conjugated bilirubin level determined. Cholestasis in a newborn can be due to infectious, genetic, metabolic, or undefined abnormalities giving rise to mechanical obstruction of bile flow or to functional impairment of hepatic excretory function and bile secretion (see Table 347-3). Mechanical lesions include stricture or obstruction of the common bile duct; biliary atresia is the prototypic obstructive abnormality. Functional impairment of bile secretion can result from congenital defects or damage to liver cells or to the biliary secretory apparatus.
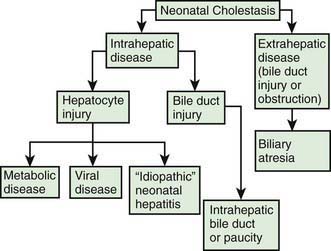
Neonatal cholestasis can be divided into extrahepatic and intrahepatic disease (Fig. 348-1). The clinical features of any form of cholestasis are similar. In an affected neonate, the diagnosis of certain entities, such as galactosemia, sepsis, or hypothyroidism, is relatively simple and a part of most neonatal screening programs. In most cases, the cause of cholestasis is more obscure. Differentiation among biliary atresia, idiopathic neonatal hepatitis, and intrahepatic cholestasis is particularly difficult.
Mechanisms
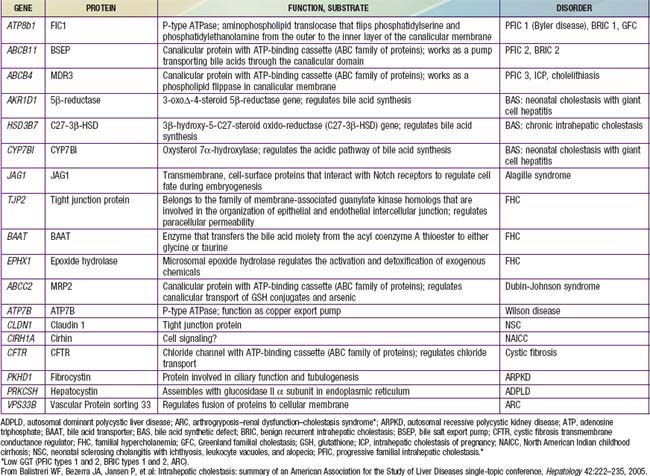
Functional abnormalities in the generation of bile flow can also have a role in neonatal cholestasis. Bile flow is directly dependent on effective hepatic bile acid excretion by the hepatocytes. During the phase of relatively inefficient liver cell transport and metabolism of bile acids in early life, minor degrees of hepatic injury can further decrease bile flow and lead to production of atypical and potentially toxic bile acids. Selective impairment of a single step in the series of events involved in hepatic excretion produces the full expression of a cholestatic syndrome. Specific defects in bile acid synthesis are found in infants with various forms of intrahepatic cholestasis (Table 348-1). Severe forms of familial cholestasis have been associated with neonatal hemochromatosis and an aberration in the contractile proteins that compose the cytoskeleton of the hepatocyte. Neonatal hemochromatosis can also be an alloimmune-mediated gestational (maternal antibodies against fetal hepatocytes) disease responsive to maternal intravenous immunoglobulin (IVIG). Sepsis is known to cause cholestasis, presumably mediated by an endotoxin produced by Escherichia coli.
Table 348-1 PROPOSED SUBTYPES OF INTRAHEPATIC CHOLESTASIS
Note: FIC1 deficiency, BSEP deficiency, and some of the disorders of bile acid biosynthesis are characterized clinically by low levels of serum GGT despite the presence of cholestasis. In all other disorders listed, the serum GGT level is elevated.
ADPLD, autosomal dominant polycystic liver disease (cysts in liver only); ARPKD, autosomal recessive polycystic kidney disease (cysts in liver and kidney); BAAT, bile acid transporter; BRIC, benign recurrent intrahepatic cholestasis; BSEP, bile salt export pump in; GGT, γ-glutamyl transpeptidase; PFIC, progressive familial intrahepatic cholestasis.
From Balistreri WF, Bezerra JA, Jansen P, et al: Intrahepatic cholestasis: summary of an American Association for the Study of Liver Diseases single-topic conference, Hepatology 42:222–235, 2005.
Evaluation
The evaluation of the infant with jaundice should follow a logical, cost-effective sequence in a multistep process (Table 348-2). Although cholestasis in the neonate may be the initial manifestation of numerous and potentially serious disorders, the clinical manifestations are usually similar and provide very few clues about etiology. Affected infants have icterus, dark urine, light or acholic stools, and hepatomegaly, all resulting from decreased bile flow due to either hepatocyte injury or bile duct obstruction. Hepatic synthetic dysfunction can lead to hypoprothrombinemia and bleeding. Administration of vitamin K should be included in the initial treatment of cholestatic infants to prevent hemorrhage.
Table 348-2 VALUE OF SPECIFIC TESTS IN THE EVALUATION OF PATIENTS WITH SUSPECTED NEONATAL CHOLESTASIS
| TEST | RATIONALE |
|---|---|
| Serum bilirubin fractionation (i.e., assessment of the serum level of conjugated bilirubin) | Indicates cholestasis |
| Assessment of stool color (does the baby have pigmented or acholic stools?) | Indicates bile flow into intestine |
| Urine and serum bile acids measurement | Confirms cholestasis; might indicate inborn error of bile acid biosynthesis |
| Hepatic synthetic function (albumin, coagulation profile) | Indicates severity of hepatic dysfunction |
| α1-Antitrypsin phenotype | Suggests (or excludes) PiZZ |
| Thyroxine and TSH | Suggests (or excludes) endocrinopathy |
| Sweat chloride and mutation analysis | Suggests (or excludes) cystic fibrosis |
| Urine and serum amino acids and urine reducing substances | Suggests (or excludes) metabolic liver disease |
| Ultrasonography | Suggests (or excludes) choledochal cyst; might detect the triangular cord (TC) sign, suggesting biliary atresia |
| Hepatobiliary scintigraphy | Documents bile duct patency or obstruction |
| Liver biopsy | Distinguishes biliary atresia; suggests alternative diagnosis |
PiZZ, protease inhibitor ZZ phenotype; TSH, thyroid-stimulating hormone.
Intrahepatic Cholestasis
Neonatal Hepatitis
The term neonatal hepatitis implies intrahepatic cholestasis (see Fig. 348-1), which has various forms (Tables 348-1 and 348-3).
Zellweger (cerebrohepatorenal) syndrome is a rare autosomal recessive genetic disorder marked by progressive degeneration of the liver and kidneys (Chapter 80.2). The incidence is estimated to be 1/100,000 births; the disease is usually fatal in 6-12 mo. Affected infants have severe, generalized hypotonia and markedly impaired neurologic function with psychomotor retardation. Patients have an abnormal head shape and unusual facies, hepatomegaly, renal cortical cysts, stippled calcifications of the patellas and greater trochanter, and ocular abnormalities. Hepatic cells on ultrastructural examination show an absence of peroxisomes. MRI performed in the 3rd trimester can allow analysis of cerebral gyration and myelination, facilitating the prenatal diagnosis of Zellweger syndrome.
Disorders of Transport, Secretion, Conjugation, and Biosynthesis of Bile Acids
PFIC 1 (FIC-1 deficiency) has been mapped to chromosome 18q12 and results from defect in the gene for F1C1 (ATP8B1; Tables 348-3 and 348-4). F1C1 is a P-type adenosine triphosphatase (ATPase) that functions as aminophospholipid flippase, facilitating the transfer of phosphatidyl serine and phosphatidyl ethanolamine from the outer to inner hemileaflet of the cellular membrane. F1C1 might also play a role in intestinal bile acid absorption, as suggested by the high level of expression in the intestine. Defective F1C1 might also result in another form of intrahepatic cholestasis: benign recurrent intrahepatic cholestasis (BRIC) type I. The disease is characterized by recurrent bouts of cholestasis, jaundice, and severe pruritus lasting from a 2 wk to 6 mo period; it can last up to 5 yr. The episodes vary from few episodes per year to 1 episode per decade and can profoundly affect the quality of life. Nonsense, frame shift, and deletional mutations cause PFIC type I; missense and split type mutations result in BRIC type I. Typically, patients with BRIC type I have normal cholesterol and GGT levels.



