Cardiovascular Disease in the Newborn
Anne Marie Valente
Craig E. Fleishman
Norman S. Talner
The clinical approach to cardiovascular problems of the newborn must take into account the structural and functional basis of normal and abnormal cardiovascular development. This includes fundamental cardiac embryology, now focused on the molecular events that control cardiac myocyte differentiation and growth; basic ultra structural and functional differences between fetal, neonatal, and adult cardiac muscle; age-related responses to imposed circulatory loads; the transition from the fetal to the neonatal circulation (particularly the role of fetal flow pathways); and the various physiologic types of impaired cardiovascular performance that may be encountered.
This chapter encompasses etiology, the essentials of the development of cardiac structure and function, elements of the fetal and neonatal circulations that may affect disease states, the clinical assessment of cardiovascular performance in the neonate, diagnostic tests that permit the recognition and accurate delineation of a potential cardiac problem, and a functional approach to diagnosis and management that takes into account special clinical problems such as congestive heart failure, impaired systemic perfusion, hypoxemia, and disturbances of cardiac rhythm.
ETIOLOGY
Most cardiovascular diseases encountered in the newborn are congenital and, as such, they represent altered structural development. This can be either abnormal development of a normal structure (aortic or pulmonary atresia, septal defects), failure of a structure(s) to progress beyond a particular embryonic stage (double-outlet right ventricle, truncus arteriosus), or modification of normal flow pathways (coarctation).
In addition to congenital heart lesions, cardiac function in the neonate may be compromised by serious rhythm disturbances (tachycardia, heart block), inflammatory diseases of the myocardium, metabolic defects (e.g., glycogen storage disease, carnitine deficiency), defects of mitochondrial electron transport that interfere with the contractile process, and the adverse effects of intrauterine asphyxia on the myocardium, atrioventricular (AV) valve function, and pulmonary circulation.
Cardiovascular malformations often accompany chromosomal disorders such as trisomy 21, or are observed in the context of certain genetically determined syndromes, metabolic defects, abnormalities of cardiac rhythm, connective tissue disorders, and abnormal tissue growth. The prognosis in many of these syndromes is determined by the cardiovascular status; thus, detailed evaluation during the newborn period is required.
Congenital heart malformations usually result from the interaction between genetic and environmental systems. In certain conditions, a specific etiology has been identified. Maternal rubella during early gestation, for example, produces a syndrome, now largely preventable, in which a patent ductus arteriosus (PDA) and pulmonary artery stenosis are common cardiovascular findings. Cardiac defects, particularly the ventricular septal defect, occur in almost 50% of infants with the fetal alcohol syndrome. The finding of complete heart block in a newborn has been linked to the presence of or subsequent development of a rheumatologic disorder in the mother. Common cardiac findings associated with some specific syndromes are listed in Chapter 52, Epidemiology of Congenital Heart Disease.
Counseling on the incidence and recurrence rates for families of infants with congenital heart disease is extremely important. The availability of fetal cardiac ultrasound studies now permits an accurate determination of the presence or absence of significant structural heart disease in subsequent pregnancies and is recommended for families in which a previous child has been born with a congenital heart defect.
NORMAL CARDIAC DEVELOPMENT
The heart is the first organ to form in vertebrates. At about embryonic day 20 in humans, cells in the anterior lateral plate mesoderm become committed to developing into cardiac cells that form a bilateral cardiogenic field. This cardiogenic field then develops into parallel cardiac primordia that fuse in the midline to form the primitive cardiac tube. The families of signaling molecules, including FGF and BMP, that lead to formation of the primitive heart are expressed by the adjacent endoderm. The myocardium initially forms a trough around the endocardial tube (Fig. 53.1). Between the myocardium and endocardium is the cardiac jelly, which is made up of glycoconjugates. Inductive interactions between the layers is necessary for several steps in heart development. The end result is the formation of a centrally located heart, from cardiac precursors that were bilateral in location. The single cardiac tube has dilatations that constitute the sinus venosus, common atrium, presumptive left ventricle, presumptive right ventricle, and the conus. The truncus is not present in the early heart tube, but is added to the end of the tube from a secondary heart field (Fig. 53.2). A constriction separates each of these regions, and these indentations define the sinoatrial segment, the AV canal, and the bulboventricular canal.
In early development, looping of the bulboventricular region occurs, which tends to move the presumptive right ventricle anteriorly and to the right of the presumptive left ventricle. Cardiac looping is the first indication of right–left asymmetry in the embryo, and it is thought to possibly occur because of differential cell growth or altered cell adhesion. Looping of the heart to the right occurs in all vertebrates, suggesting that this is a critical step in cardiac morphogenesis. Studies of mouse mutants and chick embryos have led to insights into the signals that initiate cardiac looping. At the stage of looping, the embryonic systemic venous return is to the sinus venosus, then to a common atrium and then the presumptive right ventricle via the AV canal, and, finally, to the truncus. The embryonic
heart begins to beat concomitantly with the formation of the heart tube and the onset of looping.
heart begins to beat concomitantly with the formation of the heart tube and the onset of looping.
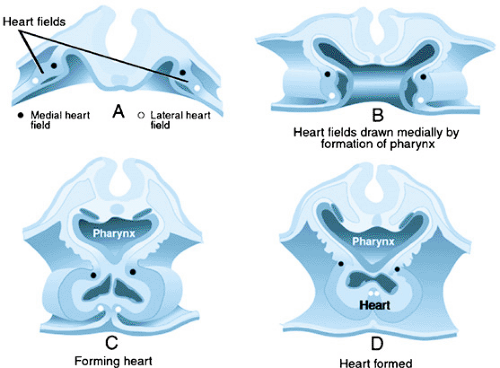 FIGURE 53.1. Diagrams showing steps in development of the bilateral primary heart fields in the splanchnic mesoderm into a single midline primary heart tube (A–D). Formation of the midline primary heart tube depends on formation of the foregut pocket, which forms the pharynx. (Reprinted with permission from Kirby ML and Waldo KL. Molecular embryogenesis of the heart. Pediatr Pathol 2002:516.) |
The looping process also confers two curvatures to the heart, with the outer curvature being more extensive than the inner. The inner curvature separates the AV canal—the locus for the developing AV valves—from the bulbar-truncal region, where the semilunar valves will originate. The semilunar valves form from endocardial cushions that line the cardiac tube distal to the AV junction. During development, the endocardial cushion tissue divides the heart into separate flow channels that prevents backflow. The remainder of the early developmental process will result in the partitioning of the atria, ventricles, conus, and truncus, and the establishment of the functioning fetal circulation by the eighth week of gestation. Molecular studies suggest that chamber specification precedes the actual septation of the heart.
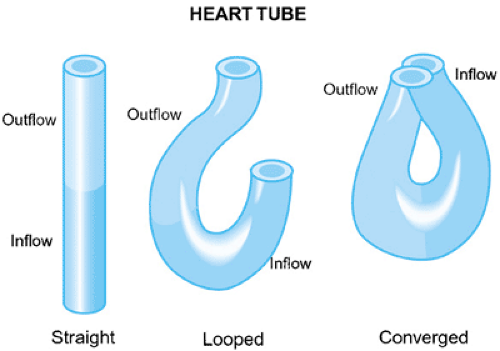 FIGURE 53.2. The primary heart tube begins as a straight midline tube with blood entering caudally via the inflow and exiting cranially via the outflow tract. Under normal circumstances, the tube loops to the right, creating an inflow limb and an outflow limb. The distal extremities of the inflow and outflow limbs grow toward each other in a process of convergence that is necessary before septation can create a four-chambered heart. (Reprinted with permission from Kirby ML and Waldo KL. Molecular embryogenesis of the heart. Pediatr Pathol 2002:516.) |
The development of the cardiac conduction system occurs prior to septation. The sinoatrial (SA) node, which is the pacemaker of the heart, forms from a caudal region of the straight heart tube. Initially, electrical impulses spread from the SA node throughout the ventricle and across the AV canal region to the ventricles. As the atria and ventricles become electrically isolated, the AV node in a region of the interventricular septum becomes the only pathway of depolarization from the atria to the ventricles. Depolarization of the ventricles continues through specialized conduction tissue. The persistence of conduction pathways around the AV fibrous ring may result in the episodic occurrence of arrhythmias.
ABNORMAL CARDIAC DEVELOPMENT
Cardiovascular morphogenesis, as described in the preceding section, involves a number of important developmental mechanisms, including cell growth, migration, death, differentiation, and adhesion. Hemodynamic factors add another component to cardiovascular development. Structural defects found in clinical settings represent those changes in cardiovascular development that are compatible with the fetal circulation. Structural defects may result from various mechanisms. The addition of myocardium from the secondary heart field, which serves to lengthen the primary heart tube, is an essential step in the alignment of the inflow and outflow tracts prior to septation. If this lengthening does not occur normally, ventricular septal defects and dextroposition of the aorta occur. A dextroposed aorta occurs in tetralogy of Fallot and double-outlet right ventricle. The genetic pathogenesis of dextroposed aorta involves proteins such as Nkx2.5, neurotrophin 3, and probably a plethora of others.
Neural crest tissue has been shown to provide ectomesenchyme not only to the pharynx, but also to the outflow tract. The same region of the neural crest provides the postganglionic innervation of the heart. Abnormalities in the migration of ectomesenchymal tissue, as demonstrated by the ablation of the premigratory neural crest in chick embryos and in patients with the 22q11,2 deletion Syndrome (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, and hypocalcemia resulting from microdeletions of chromosome 22q11), results in cardiac malformations. Some of these malformations include interruption of the aortic arch, persistence of the truncus arteriosus, and double-outlet right ventricle. Development of the parathyroid gland, thymus, and conotruncal region has been linked to neural crest function, and can account partially for the phenotype in DiGeorge syndrome. Investigations of zebrafish and mouse models seek to better define the genetic basis of many of these malformations.
Abnormalities of intracardiac blood flow can result in left and right heart obstructive defects, such as semilunar valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome. Abnormal targeted growth can result in anomalous pulmonary venous return. Abnormalities of cell death lead to such lesions as Ebstein malformation of the tricuspid valve and muscular ventricular septal defects. Ventricular growth is particularly sensitive to genetic mutations. In mice models, multiple mutations of genes result in hypoplasia of the ventricular wall and ventricular septal defects.
Extracellular matrix abnormalities are associated with atrioventricular canal defects (AVC) and dysplastic semilunar valves. Investigations into molecular pathways have shown that the normal migration of cells in the extracellular matrix is dependent on the variable expression of certain adhesion molecules throughout the heart (e.g., neural cell adhesion molecule; tenascin). Changes in the normal migration of cells may cause the above-mentioned lesions. The high prevalence of AV canal defects in children with Down syndrome has contributed to the awareness of identifying the critical gene(s) on chromosome 21 that may be linked to the cardiac defects. Down syndrome cell adhesion molecule (DSCAM) is expressed during cardiac development, and increased adhesiveness of mesenchymal cells in the endocardial cushions has been proposed as one of the pathologic mechanisms that contribute to cardiac defects. In addition, the absence of a cofactor essential for the normal expression of myocardial genes will result in a common AVC.
Situs and looping defects result in heterotaxy syndrome, in which a reversal or loss of visceral asymmetry occurs. This syndrome almost always is associated with complex structural defects in the heart. Right–left axis formation is dependent on ciliary movements through a microtubule-dependent motor protein. Activin also plays a role in right–left axis formation. Mice that are deficient in activin receptor IIB display randomized heart looping and malposition of the great arteries.
The normal development of the great arteries involves the transformation from a paired six-aortic-arch system into a definitive vascular pattern, as shown in Figure 53.3. Anomalies of the aortic arch artery derivatives are illustrated in Figure 53.4.
Airway Obstruction with Vascular Malformations
A vascular ring is formed when the trachea and esophagus are completely surrounded by vascular structures. Clinical manifestations of vascular rings range from severe respiratory difficulty to asymptomatic individuals in whom the abnormality is discovered at the time of chest imaging or autopsy. Symptoms depend upon the tightness of the ring. Infants with vascular rings may have significant stridor and hyperextension of their necks. A history of frequent upper respiratory infections, bronchiolitis, or pneumonia is common. Infants who have a history of “noisy breathing since birth” that significantly worsens with upper respiratory tract infections should raise the suspicion of
a vascular ring. Dysphagia secondary to aortic arch abnormalities is a rare finding in infants.
a vascular ring. Dysphagia secondary to aortic arch abnormalities is a rare finding in infants.
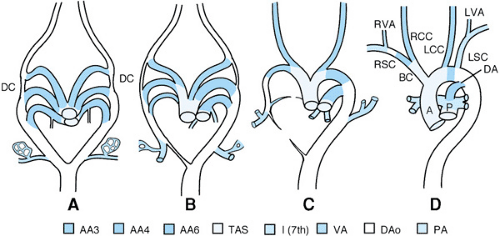 FIGURE 53.3. Schematic summary of the transformation of the symmetrical early human pharyngeal arterial basket into the configuration of the great arteries at birth. A: The pattern of paired arch arteries is symmetrical as they branch from the truncoaortic sac and join the dorsal aorta. The first and second arches have disappeared, leaving the third, fourth, and sixth arch arteries. The seventh intersegmental arteries branch from the dorsal aorta cranial to the aortic bifurcation. B and C: Transition stages between the early pharyngeal arterial pattern and the final configuration of the great arteries. The right ductus arteriosus narrows and disappears, and the left ductus and the primitive pulmonary arteries enlarge. The dorsal aorta on the left side enlarges, whereas the dorsal aorta on the right remains smaller in diameter. The aortic sac elongates causing the fourth arch arteries to shorten and become transported cranially. D: The configuration of the great arteries at birth. Reprinted with permission from Waldo K and Kirby ML. In: Markwald R, ed. Living morphogenesis of the heart. Boston: Birkhauser, 1999.) |
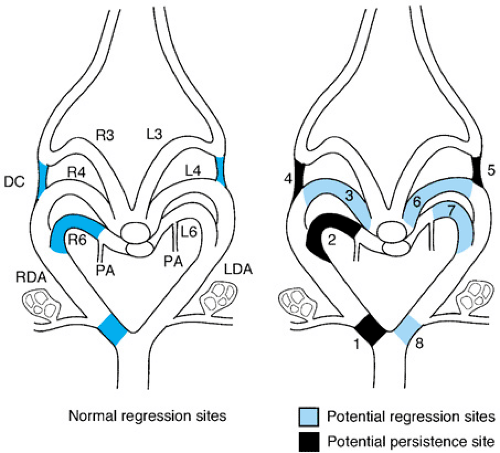 FIGURE 53.4. The drawing on the left illustrates the normal sites of vessel regression to produce normally patterned great arteries and the drawing on the right shows the potential sites of regression that are seen in human malformations involving abnormal patterning of the great arteries (1–8). R3, L3, right and left third arch artery; R4, L4, right and left fourth arch artery. R6, L6, right and left sixth arch artery; PA, pulmonary artery; DC, carotid duct; RDA, LDA, right and left dorsal arch. (Reprinted with permission from Waldo K and Kirby ML. In: Markwald R, ed. Living morphogenesis of the heart. Boston: Birkhauser, 1999.) |
To understand the vascular ring, it is necessary to appreciate the embryologic development of the aortic arch arteries, as described in the section, Normal Cardiac Development (Fig. 53.4). Additionally, the identification of the sidedness (right or left) of the definitive aortic arch is particularly important. The normal aortic arch is leftward. A right aortic arch may be found in the setting of a structurally normal heart; however, a significant incidence of right aortic arch occurs with cardiac lesions such as tetralogy of Fallot and persistent truncus arteriosus.
Double Aortic Arch
Double aortic arch has the presence of both a right and left aortic arch due to persistence of both fourth aortic arch arteries. In this situation, one arch can be either hypoplastic or atretic; this is usually the left arch. Alternatively, both arches may be patent, in which case infants are usually symptomatic with stridor during the first several weeks of life.
Right Aortic Arch Anomalies
A right aortic arch with a diverticulum of Kommerell occurs when there is persistence of the left sixth aortic arch artery (ductus) between the truncoaortic sac and left dorsal aorta, and dissolution of the left fourth aortic arch artery. This results in the left carotid artery as the first branch off the aortic arch, followed by the right carotid artery, and right subclavian artery, and a retroesophageal vessel that gives rise to the left subclavian artery and a left ductus or ligamentum. This particular anomaly can be difficult to distinguish from a right aortic arch with retroesophageal subclavian artery, although the diverticulum of Kommerell is usually a significantly larger vessel than the subclavian artery. Classically, vascular rings were diagnosed by barium swallow esophograms, however these have been largely replaced by ultrasound, computerized tomography and magnetic resonance imaging. Most children with this arch anomaly are asymptomatic, and surgical correction for the rare symptomatic patient is done most often through a left thoracotomy. A rare variation of the right aortic arch exists in which the left ductus or ligamentum arises from a retroesophageal diverticulum from the right-sided descending aorta, and this can produce respiratory symptomatology.
A right aortic arch with left descending aorta and left ductus arteriosus or ligamentum is the result of the disappearance of either the left fourth aortic arch artery or left dorsal aorta distal to the takeoff of the left subclavian artery. The vascular ring is produced by persistence of the left sixth aortic arch artery as it connects to the left-sided dorsal aorta. These vascular rings usually are nonobstructive, and surgical correction is indicated only when the child is symptomatic.
A right aortic arch with retroesophageal innominate artery is another rare anomaly in which the right carotid artery is the first arch vessel, followed by the right subclavian artery and then the retroesophageal left innominate artery. The ductus or ligamentum connects the left pulmonary artery with the base of the innominate artery and forms the vascular ring. This malformation often is suspected through imaging that reveals a single carotid artery arising as the first branch from the proximal aorta.
Left Aortic Arch Anomalies
Left aortic arch with right descending aorta and right ductus or ligamentum is an arch anomaly in which persistence occurs of the right sixth aortic arch artery, which connects to the right pulmonary artery. The surgical approach to this lesion differs from most vascular rings, because a right thoracotomy is preferred to visualize and divide the ligamentum.
Cervical Aortic Arch
A cervical aortic arch is found above the level of the clavicle. A vascular ring exists when an anomalous subclavian artery is present. This usually is associated with a right aortic arch that descends on the right to about the level of the T4 vertebrae, where it crosses behind the esophagus to the left and gives off the left subclavian artery and a ductus or ligamentum. Presentation of cervical arches in infancy may be pulsatile masses in the neck or supraclavicular fossa. Femoral pulses diminish with compression of the mass. Infants have typical symptoms of a vascular ring, including stridor, recurrent respiratory infections, and respiratory distress. Chest roentgenogram may reveal a widened mediastinum without an evident aortic knob.
Pulmonary Artery Slings
In this rare anomaly, the lower portion of the trachea is partially surrounded by vascular structures, with the left pulmonary artery arising from the right pulmonary artery. It is the only instance in which a major vascular structure courses between the trachea and the esophagus. This anomaly often is associated with complete cartilaginous rings in the distal trachea, which can lead to significant tracheal stenosis. Infants present with respiratory distress and stridor. Barium esophagraphy classically reveals anterior indentation of the esophagus. The traditional surgical approach has been a division of the left pulmonary artery from the right pulmonary artery and reanastomosis of the left pulmonary artery in front of the trachea. However, a surgical approach has been described that involves transecting the trachea and reanastomosing it posterior to the pulmonary artery bifurcation, which permits tracheal reconstruction if cartilaginous rings are present.
Innominate Artery Syndrome
An anterior compression of the trachea by the innominate artery may occur, although this is not a form of a vascular ring. Previously, this was thought to be due to a more distal and leftward takeoff of the innominate artery, although imaging studies verifying this are inconsistent. Tracheomalacia often is associated with this situation, and infants present with inspiratory stridor. A lateral chest roentgenogram may reveal anterior indentation of the tracheal air column. Surgical interventions may be required to relieve the compression. These include suspension of the innominate artery from the sternum, or moving the innominate artery more proximal on the aortic arch.
Absent Pulmonary Valve Syndrome
Severe airway problems may occur with tetralogy of Fallot with an absent pulmonary valve. In utero, this lession may result in massive pulmonary regurgitation and the development of significant dilation of the main pulmonary artery and compromise of the large airways and interference with lung development.
DEVELOPMENT OF CARDIOVASCULAR FUNCTION
The primitive embryonic heart initially is a muscle-wrapped contractile tube without valve function, partitions, or a coronary circulation. This circulating system is capable of providing for the oxygen and nutrient needs of the embryo while at the same time removing the end-products of metabolism.
During fetal life, the placenta serves as the site for gas exchange, with almost fully oxygenated blood returning to the fetal body via the umbilical veins. The common umbilical vein enters the porta hepatis, where it is joined by the portal vein. Branches are provided to the left and right lobes of the liver, with the ductus venosus serving as a direct connection to the
inferior vena cava, thus permitting umbilical venous blood to bypass the hepatic microcirculation. The well-oxygenated umbilical stream is directed across the foramen ovale, allowing blood of the highest O2 concentration to perfuse the cerebral circulation. The third shunt channel, the ductus arteriosus, allows blood from the right ventricle to enter the descending aorta and essentially bypass the pulmonary circulation. The fetal heart and circulation with functioning flow pathways are shown in Figure 53.5.
inferior vena cava, thus permitting umbilical venous blood to bypass the hepatic microcirculation. The well-oxygenated umbilical stream is directed across the foramen ovale, allowing blood of the highest O2 concentration to perfuse the cerebral circulation. The third shunt channel, the ductus arteriosus, allows blood from the right ventricle to enter the descending aorta and essentially bypass the pulmonary circulation. The fetal heart and circulation with functioning flow pathways are shown in Figure 53.5.
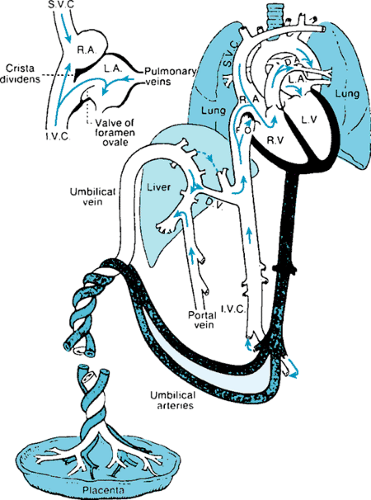 FIGURE 53.5. The fully developed fetal circulation, indicating the fetal flow pathways—foramen ovale, ductus arteriosus, and ductus venosus. The insert shows the direction of flow of the more highly oxygenated placental stream across the foramen ovale. RA, right atrium; LA, let atrium; RV, right ventricle; LV, left ventricle; DA, ductus arteriosus; FO, foramen ovale; DV, ductus venosus; IVC, inferior vena cava; SVC, superior vena cava. (Reproduced with permission from Adams FH, Emmanouilides GC, eds. Heart disease in infants, children, and adolescents, 3rd ed. Baltimore: Williams & Wilkins, 1983:12.) |
The patterns of blood flow in the fetal circulation have been studied in a fetal lamb model, using microsphere techniques. These studies have shown that about 20% of the total systemic venous return is derived from the superior vena caval stream, 3% is derived from the coronary circulation, and 7% is derived from the pulmonary vascular bed. Inferior caval flow accounts for 70% of the systemic venous return, of which 40% derives from umbilical veins, 5% from portal veins, and 25% from the lower fetal body. Fifty-five percent of umbilical venous flow passes through the ductus venosus, whereas 45% flows via the right and left lobes of the liver. Of interest, the left lobe is supplied from the umbilical vein, whereas the majority of the right lobe is perfused with portal venous blood. Under conditions of hypoxemia and flow limitation, a redistribution of venous return occurs, favoring flow through the ductus venosus.
Thus, blood at the level of the entry of the inferior vena cava into the right atrium is derived from four sources: the ductus venosus (umbilical vein), the distal inferior vena cava, the right hepatic vein, and the left hepatic vein. The most highly oxygenated stream (ductus venosus) is directed preferentially across the foramen ovale, whereas the more distal inferior vena caval blood with a much lower O2 saturation passes across the tricuspid valve. The higher saturations of left ventricular blood ensure adequate oxygenation of the developing brain as well as provide vital substrates for brain metabolism.
The superior vena caval stream is directed across the tricuspid valve, with only 2% passing from right to left across the foramen ovale. The net effect of the fetal venous return pathways is a lower O2 saturation on the right side compared with that on the left. The volume of blood in the right and left ventricles also favors the right side in the lamb. Ultrasound and Doppler-derived flow studies across the mitral and tricuspid valves in the developing human fetus, however, indicate that the volume flows on the right and left sides of the heart are almost equal, with only a slight dominance of right ventricular output (55% versus 45%).
The fetal cardiac output represents the combined output of both ventricles. Most of the right ventricular output of the fetus flows into the descending aorta via the ductus arteriosus, whereas the majority of the left ventricular output perfuses the upper body with the aortic isthmus (a segment from the origin of the left subclavian artery to the ductus arteriosus) receiving only 30% of blood from the ascending aorta. The isthmus is the site of the functional separation of the two circuits.
We turn next to an evaluation of fetal and neonatal cardiovascular performance. When compared to the adult heart, the fetal and newborn myocardium is unique with respect to its ultrastructure, mechanical and biochemical properties, metabolism, and autonomic innervation. The developing cardiac myocyte is smaller than the adult cardiac myocyte, and contains relatively more noncontractile mass, with a reduction in cell density. Consequently, force generation and the extent and velocity of shortening are decreased, whereas water content and stiffness are increased. The process of excitation–contraction coupling also differs in the fetus and newborn, when compared with that in the adult. Less development of the sarcoplasmic reticulum and T tubule system is present in the fetus, which may interfere with the movement of calcium ions. Calcium ion increase in the cytosol appears to be provided by trans-sarcolemmal movement rather than by the release of calcium from the sarcoplasmic reticulum, as in the adult. Myofibrillar adenosine triphosphatase levels are low, and age-related differences in contractility also may occur as the result of the decreased activation of the modulatory proteins, troponin and tropomyosin.
Functional integrity exists for the afferent and efferent limbs of the autonomic pathways, although adrenergic innervation is incomplete in the fetus and newborn. This implies that circulating vasoactive agents may play a greater role in cardiovascular adaptation in the more immature individual.
The response of the fetal and newborn myocardium to altered loading conditions reflects diminished cardiovascular reserve, although this point is somewhat controversial. A limited ability may be present to increase cardiac output in response to a volume load, yet the Frank-Starling mechanism is operative. The tension developed during ejection thereby results in a limited increase in stroke volume. In some studies, inotropic interventions produce less of an augmentation in heart rate and cardiac output than is seen in older age groups, whereas decreases in heart rate (to less than 100 beats per minute) appear to be associated with pronounced falls in cardiac output. The fetus and newborn can modify the distribution of cardiac output by increasing vasomotor tone. This is a major adaptive mechanism that permits the perfusion of the myocardium and brain at the expense of the peripheral circulation.
It should be stressed that the overall response of the heart and circulation to a specific stimulus (e.g., volume, afterload increase) is an integrated response. For example, when afterload is increased by increasing systemic vascular resistance, a preload effect occurs as ventricular volume increases. Furthermore, if volume is increased, blood pressure will rise, and thus be associated with a change in afterload. The factors that may be involved in the integrated response of the heart and circulation include: (a) diastolic or filling volume; (b) ejection tension (afterload); (c) vascular impedance; (d) heart rate; (e) contractile state; (f) compliance; and, (g) ventricular interdependence (cross talk). The latter is especially important in the fetus and newborn, in whom volume loading of one ventricle may modify the performance characteristics of the other ventricle. For example, a volume-loaded right ventricle alters the filling characteristics of the left ventricle and vice versa.
Oxygen delivery to the myocardium in the fetus is accomplished through a coronary blood flow per gram that is 90% higher than that in the adult. Myocardial metabolism is maintained via the glycolytic pathway, with nearly 60% of fetal myocardial energy requirements being met by lactate extraction through the tricarboxylic acid cycle. Free fatty acids, the dominant energy source in the adult, are not used as a substrate for the fetal heart. L-Carnitine, a required cofactor for the beta-oxidation of fats, is present in low concentrations in the immediate postnatal period.
As oxygen tension rises after birth, the rate of oxidative metabolism increases and the mass of mitochondria per gram of myocardial tissue doubles. Cytochromes c1, a1 and a3, and cytochrome oxidase rise in concert with the increase in oxygen tension. The myocardial isoenzyme pattern changes from the anaerobic muscle form to the aerobic form, reflecting the transition from the intrauterine to the extrauterine environment.
EFFECTS OF CARDIAC MALFORMATION ON FETAL CIRCULATORY FUNCTION
In most instances, cardiac malformations are tolerated well by the developing fetus because of the presence of the fetal flow pathways (which permit the bypass of obstructive lesions), the existence of the placenta for gas exchange, and the high pulmonary vascular resistance. On occasion, however, the presence of certain malformations interferes with the growth of chamber size, such as when a small foramen ovale compromises left heart development, or when incorporation of the common pulmonary vein into the left atrium fails, resulting in a hypoplastic left atrium (total anomalous pulmonary venous connection). Dominance of one ventricle over the other can modify the size of the great arteries, with the aortic isthmus becoming hypoplastic when flow from the pulmonary artery to the ductus into the descending aorta is the predominant flow pattern. When pulmonary atresia or severe obstruction of right ventricular outflow is present, the preferred pathway is via the aorta, which tends to be enlarged, whereas the pulmonary artery(s) is hypoplastic.
In a few congenital heart defects, the fetal cardiac function is compromised, with cardiac failure arising in utero. This occurs when massive AV valve regurgitation, semilunar valve insufficiency, or a large systemic arteriovenous fistula (vein of Galen malformation, giant hemangioma) is present. These share the common hemodynamic theme of right-sided volume overloading, either primarily or secondarily, when left-sided lesions are present. Hydrops fetalis with pericardial and pleural effusions, ascites, and generalized anasarca may occur in the face of these disturbances. This clinical picture also may develop with prolonged fetal tachyarrhythmias. These rhythm disturbances are being managed in some instances by the administration of antiarrhythmic pharmacologic agents (propranolol, digoxin, procainamide) to the mother, with transplacental passage of the agent to the fetus to enable control of the arrhythmia and resolution of the hydrops. Thus, intrauterine diagnosis is the key to successful management. Massive cardiomegaly, which can be observed in association with fetal hydrops, also may compromise lung development, an observation that is similar to that which takes place with diaphragmatic hernias.
DEVELOPMENT OF THE PULMONARY CIRCULATION
The pulmonary arterial tree develops in conjunction with the airways. The periacinal, arterial, and airway branching pattern is completed by the sixteenth week of gestation. The intra-acinar vessels and alveoli develop during the latter part of gestation and postnatally. The multiplication of intra-acinar arteries keeps pace with alveolar ducts and alveolar development, and is particularly rapid during the first years of life. This progressive growth and remodeling of the pulmonary vascular bed provides sufficient capacity to ensure a low pulmonary vascular resistance.
During fetal life, the resistance vessels (precapillary) have a thicker muscular wall than the vessels in a child or adult. This reflects the high pulmonary vascular resistance of the fetus (increased pressure drop across the bed and low pulmonary blood flow). In the postnatal remodeling process, a progressive decrease occurs in muscularity as pulmonary vascular resistance falls. The important factors to be taken into consideration in the assessment of pulmonary vascular resistance are the pressure drop from the pulmonary arteries to the veins, pulmonary blood flow, surface area, muscularity and tone of the resistance vessels, and blood viscosity. The postnatal fall in hematocrit results in a decrease in blood viscosity and contributes to the lowering of pulmonary vascular resistance. The presence of pulmonary hypertension does not necessarily imply an increase in pulmonary vascular resistance if pulmonary blood flow is increased markedly, as occurs with large-volume left-to-right shunts.
In infants with persistent pulmonary hypertension of multiple etiologies, the pulmonary artery pressure is close to or at systemic levels, whereas pulmonary blood flow is decreased (high resistance state). It is imperative, therefore, in the assessment of cardiopulmonary disorders of the infant that the resistance of the pulmonary circulation be taken into consideration. Furthermore, persistent pulmonary hypertension is not in itself a definitive diagnosis. A precise diagnosis is required of the cause of pulmonary hypertension (e.g., lung disease, cardiac problem, primary disorder of pulmonary resistance vessels) so that appropriate management can be carried out. Echocardiography occupies a key role in defining the specific nature of the problem in the pulmonary circulation.
OXYGEN TRANSPORT IN THE FETUS AND NEWBORN
Oxygen transport (delivery) represents the product of the cardiac output and the arterial oxygen content. The arterial oxygen content is determined by the oxygen tension, the affinity of hemoglobin for oxygen, and the absolute hemoglobin level. The hemoglobin concentration is controlled by erythropoietin production. The liver is the site of erythropoietin production in the fetus, as contrasted with erythropoietin production by the kidney in the adult. Because the fetus is exposed to a lower oxygen saturation, the erythropoietin response differs
qualitatively or has a different sensitivity to hypoxemia than does the response at later stages of development. The affinity of hemoglobin to oxygen is greater in the fetus and newborn. This is accounted for by the fact that fetal hemoglobin has less affinity for 2,3-DPG (diphosphoglycerate), which competitively decreases hemoglobin–oxygen affinity. This is important for oxygen uptake in the fetus, because placental oxygen transfer occurs at much lower oxygen tension than does alveolar oxygen transfer. The P50 (PO2 at which hemoglobin is 50% saturated with oxygen) is 20 mm Hg for fetal blood and about 28 mm Hg for adult blood. Therefore, postnatally, with an arterial PO2 of 100 mm Hg and a venous PO2 of 40 mm Hg, the amount of oxygen extracted from fetal blood is much less than that extracted from adult blood.
qualitatively or has a different sensitivity to hypoxemia than does the response at later stages of development. The affinity of hemoglobin to oxygen is greater in the fetus and newborn. This is accounted for by the fact that fetal hemoglobin has less affinity for 2,3-DPG (diphosphoglycerate), which competitively decreases hemoglobin–oxygen affinity. This is important for oxygen uptake in the fetus, because placental oxygen transfer occurs at much lower oxygen tension than does alveolar oxygen transfer. The P50 (PO2 at which hemoglobin is 50% saturated with oxygen) is 20 mm Hg for fetal blood and about 28 mm Hg for adult blood. Therefore, postnatally, with an arterial PO2 of 100 mm Hg and a venous PO2 of 40 mm Hg, the amount of oxygen extracted from fetal blood is much less than that extracted from adult blood.
THE TRANSITIONAL CIRCULATION
At birth, the critical event in the transition from the intrauterine to the extrauterine state is the establishment of lung inflation. This initiates a series of events, culminating in a fall in pulmonary vascular resistance and functional closure of the fetal flow pathways. The contact of air containing oxygen with the pulmonary resistance vessels produces vasodilatation, which is accompanied by a marked increase in pulmonary blood flow as pulmonary vascular resistance falls acutely. A continuing search has gone on for the specific mediator of the pulmonary vasodilatation that occurs at birth. Initial focus was on oxygen effect and bradykinin, then on prostaglandins, and, most recently, on the endothelial-derived relaxing factor, nitric oxide (NO). This has important clinical implications in the management of persistent pulmonary hypertension.
While pulmonary vascular resistance is falling, systemic vascular resistance rises consequent to the obliteration of the low-resistance placental circuit. The ductus venosus flow channel also is eliminated at this time.
Left atrial pressure rises as pulmonary venous return increases, and this produces functional closure of the foramen ovale. The postnatal rise in arterial oxygen tension, together with local changes in prostaglandin metabolism, produces a constriction of the ductus arteriosus, with functional obliteration usually complete in a full-term infant by 10 to 15 hours of life. In a preterm infant, however, the mechanisms responsible for ductal constriction are incompletely developed, and closure is delayed.
Any interference with the establishment of ventilatory function (i.e., birth asphyxia) tends to maintain the circulation in the fetal state, with a high pulmonary vascular resistance and right-to-left shunting via the ductus arteriosus and foramen ovale. This can produce the clinical picture associated with persistent pulmonary hypertension, as will be discussed later.
The final obliteration of the fetal flow pathways produces the adult-type circulation that is present in the term infant within a few weeks of birth. The two ventricles now operate in series, with a relatively high cardiac output resulting from the demands of growth and control of body temperature. Pulmonary vascular resistance is almost at adult levels, with resistance vessels characterized by very little medial thickness and increased lumen diameter. Remodeling of the pulmonary vascular bed continues postnatally, however, in concert with airway development.
KEY POINTS IN CLINICAL EVALUATION
As technology advances, pediatricians must continue to attain exceptional skills at taking a history and performing the physical examination, because this initial assessment often provides invaluable information regarding the functional cardiac diagnosis and leads to a specific etiology and necessary medical and surgical management.
History
The principal points in the history that aid in the clinical evaluation of the potentially critically ill infant with cardiovascular disease include the birth history, the presence of a chromosomal abnormality, a sibling with congenital heart disease, hydrops fetalis and ectopy, intrauterine infections, and other major-organ systemic malformations. A low Apgar score should raise the possibility of the deleterious effects of hypoxemia, hypercarbia, and, acidemia on the myocardial, pulmonary, and systemic circulations. Chronic intrauterine hypoxia is associated with persistent pulmonary hypertension with right-to-left ductal and foraminal shunting. Almost all the chromosomal abnormalities are associated with a high incidence of congenital heart disease, with trisomies 21, 13, and 18 heading the list. Although defects involving the AVC are observed commonly in infants with trisomy 21, other malformations have been seen, such as tetralogy of Fallot and PDA. The American Academy of Pediatrics (AAP) Committee on Genetics recommends all infants with trisomy 21 should undergo a two-dimensional echocardiographic assessment in the routine neonatal evaluation.
Congenital heart disease in a previous child in the family should alert the clinician to possible involvement in a subsequent pregnancy. The use of fetal echocardiography is becoming increasingly important in the prenatal diagnosis of congenital heart disease. Fetal echocardiography should be considered in the following situations: (a) an extracardiac malformation is identified; (b) exposure to a potentially teratogenic agent; (c) family history of congenital heart defects; (d) suspected fetal chromosomal abnormality or genetic disease; (e) maternal disease associated with structural heart defects or fetal cardiac arrhythmia; or (f) suspected cardiac defects based on findings of routine ultrasound.
Intrauterine infections can be associated with life-threatening myocardial dysfunction. Therefore, myocardial structure and function should be assessed in infants suspected of having a septicemic process, particularly if clinical evidence exists of low systemic perfusion.
The presence of malformations that affect multiple organ systems, such as tracheoesophageal fistula, imperforate anus, and cleft palate, should warrant a search for cardiac involvement. Abnormalities of cardiac position (dextrocardia, mesocardia) also demand a cardiac evaluation, whereas a midline liver with or without an abnormality of cardiac position should raise the possibility of atrial isomerism in association with absence of the spleen or polysplenia. The presence of asplenia or polysplenia with a midline liver warrants a radiographic evaluation for malrotation anomalies of the gut. Another important group consists of patients who have aortic arch anomalies, where a strong association exists between the absence of the parathyroid gland and thymus and conotruncal malformations.
Physical Examination
Respiratory Patterns in the Newborn
The pattern of respirations may provide valuable clues to the presence of cardiovascular disease. It is critical to examine the respiratory patterns of an infant because the ausculatory examination may be unremarkable, despite the presence of significant pulmonary edema.
Tachypnea
When lung water is increased, with the accumulation of fluid in the pulmonary interstitial space, tachypnea (rapid, shallow respirations) ensues in response to the stimulation of juxtacapillary volume receptors. This response occurs under any circumstance in which a fluid leak into the pulmonary interstitial space is present (left-to-right shunting, left heart obstructive lesions, inflammatory diseases of the myocardium or lungs, delayed fluid reabsorption). Therefore, tachypnea is a nonspecific finding that demands a careful search for its underlying cause. The diagnosis of transient tachypnea, although descriptive of a breathing pattern, does not provide a precise delineation of etiology. Supporting studies are required to rule in or out cardiovascular or pulmonary problems associated with tachypnea.
Wheezing
Transudation of fluid (water) into peribronchiolar spaces increases small airway resistance and results in wheezing on a cardiovascular basis.
Hyperpnea
When the arterial oxygen tension is extremely low and pulmonary perfusion is decreased or normal, the ventilatory response is that of hyperpnea (rapid, deep respirations), in contrast to the congested lung, in which tachypnea is observed. The hyperpneic response represents hypoxemic stimulation of the arterial chemoreceptors and results in the development of a respiratory alkalosis.
Stridor
An inspiratory stridor in a newborn infant should raise the suspicion of a vascular ring or sling.
Cardiac Evaluation
Perfusion
The status of systemic perfusion must be evaluated carefully. A low output state with any one of a number of causes should be suspected when pulses are difficult to palpate, extremities are cold and pale, and capillary refill is slow. Although this could arise from overwhelming infection, it is common to encounter this clinical picture in newborn infants with critical left heart obstruction or underdevelopment, tachyarrhythmia, or myocardial disease. It should be stressed that palpable femoral pulses in a newborn do not rule out coarctation if the ductus arteriosus is open (see the section on low perfusion states). Bounding peripheral pulses are observed with aortic runoff lesions, notably patent ductus, truncus arteriosus, systemic arteriovenous fistulas, and in the face of severe anemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


