Carbohydrate Homeostasis
Edward S. Ogata
Glucose homeostasis results from the net balance between systemic organ requirements and the production and regulation of glucose. The neonate’s ability to maintain glucose homeostasis is less well developed than the older child or adult because it is in a metabolic transition period. The abrupt switch from intrauterine life, in which glucose and metabolic fuels are provided in a well-regulated manner, to a situation in which meals are intermittent and which necessitates regulation of exogenous glucose and production of endogenous glucose. As the capability to perform these functions continues to develop in the neonate, clinical disorders that can afflict the neonate may perturb this balance, resulting in hypoglycemia or hyperglycemia. In addition, antecedent intrauterine events can alter the development of glucoregulatory capabilities in the fetus, resulting in altered neonatal glucose homeostasis.
To understand the processes responsible for glucose homeostasis in the normal neonate, an appreciation of the development of glucoregulatory capabilities in the fetus is necessary. This chapter reviews this information and its relation to the clinical disorders associated with altered neonatal glucose homeostasis. Information on the perinatal aspects of diabetes in pregnancy also is presented.
MATERNAL METABOLISM DURING PREGNANCY
Although the metabolic alterations that develop in the pregnant woman throughout gestation favor the growth and development of the fetus, the first half of gestation is also a critical period for maternal anabolism. The increased calories ingested by the woman during early gestation sustain fetal growth and facilitate maternal fat deposition. This is important preparation for the second half of gestation, a period of exponential fetal growth during which these maternal stores are mobilized to meet fetal needs. The storage of maternal energy stores is facilitated by the increased secretion of insulin that occurs in women with normal carbohydrate metabolism (1,2,3,4,5).
Changes in maternal insulin secretion and action are dramatic during the second half of pregnancy. During the first half, basal insulin secretion is normal, and insulin secretion mediates the laying down of metabolic fuels (6). In the second half of gestation, insulin secretion is greatly exaggerated (7,8). Intravenous glucose tolerance testing indicates that by the third trimester, normal pregnant women secrete three times the insulin than they do in the nonpregnant state (1).
The development of insulin resistance during the second half of pregnancy is responsible for this exaggerated insulin secretion. Responsiveness to insulin develops 40% to 60% during the second half of gestation (7,8). The conceptus causes this insulin resistance in part by the generation of human placental lactogen, progesterone, and estrogen which become available in ever increasing amounts as gestation progresses (9,10,11). These directly antagonize the effect of maternal insulin. Insulin resistance increases in liver, adipose tissue, and skeletal muscle (12). Insulin binding appears to be normal during late gestation, indicating that insulin resistance is mediated by events distal to receptor/insulin binding. It is possible that tyrosine kinase inhibition may also contribute to insulin resistance (12,13).
Preexisting maternal diabetes potentiates the effects of the antiinsulin factors, causing an excessive provision of glucose and other metabolic fuels to the fetus. This is the basic perturbation responsible for the problems of the infant of the diabetic mother (Fig. 37-1) (1,14).
Metabolic fuel availability is guaranteed to the fetus even during brief maternal fasting. After an overnight fast, pregnant women have significantly lower plasma glucose concentrations than fasted nongravid women (8,15); however, glucose production in the mother is significantly increased (16,17). This increased production ensures the provision of glucose to the fetus.
Prolonged maternal fasting does alter fuel provision to the fetus. As the fast progresses, maternal ketogenesis progressively increases (15,18). The human fetal brain at early gestation can use ketones (Fig. 37-2) (19). However, this ability to use an alternative fuel may be harmful rather than beneficial to the fetus. Offspring of mothers who were ketotic during pregnancy appear to have an increased incidence of cognitive and psychomotor delay at 3 and
5 years of age (20). The mechanisms responsible for this potential association between ketone body oxidation and impaired brain function are unknown. Because of this uncertainty, maternal fasting, even foregoing breakfast, should be avoided during pregnancy.
5 years of age (20). The mechanisms responsible for this potential association between ketone body oxidation and impaired brain function are unknown. Because of this uncertainty, maternal fasting, even foregoing breakfast, should be avoided during pregnancy.
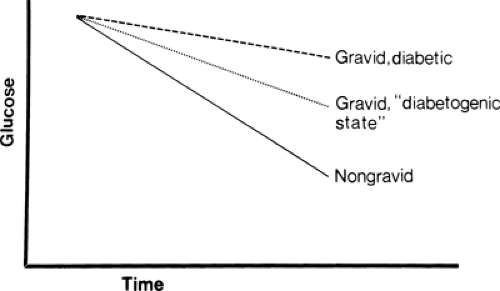 Figure 37-1 Plasma glucose changes after glucose challenge in nongravid, gravid, and gravid diabetic women. Compared to the nongravid woman, the pregnant woman with normal carbohydrate metabolism demonstrates delayed glucose clearance from midgestation onward as a result of antiinsulin factors that develop during pregnancy. The delay in glucose clearance from the maternal circulation assures glucose provision to the fetus, particularly during the postprandial period. The blunting of maternal glucose clearance is exaggerated by the counterinsulin factors in the woman with diabetes mellitus. The decreased clearance of glucose and other metabolic fuels stimulates fetal insulin production and is responsible for many of the problems of the infant of the diabetic mother. |
DEVELOPMENT OFGLUCOSE-PRODUCING AND GLUCOREGULATORYCAPABILITIES IN THE FETUS
To understand the problems of neonatal glucose homeostasis, the development of glucose production and regulatory capabilities in the fetus must be understood.
Glycogen
The third trimester of the human pregnancy is the first period in gestation during which some of the energy and substrate available to the fetus can be channeled from meeting needs for ongoing growth and development to energy storage. As the third trimester progresses, fat deposition and hepatic glycogen storage increase (21). The human fetus can synthesize and mobilize glycogen and respond to the signals that regulate these processes as early as the ninth week of gestation (22). However, only minute quantities of hepatic glycogen are present in early gestation as the great bulk of hepatic glycogen accumulates during the third trimester (22,23).
Several types of infants are at risk for neonatal hypoglycemia as a result of limited hepatic glycogen stores. Infants delivered prematurely have an abbreviated or no third trimester and thus have limited glycogen stores. Fetuses who are growth-retarded (i.e., small-for-gestational-age [SGA]) on the basis of limited metabolic fuel availability and diminished gaseous exchange (i.e., uteroplacental insufficiency) will use these fuels for growth and not for glycogen synthesis. Perinatal stress causes neonatal hypoglycemia in part because of catecholamine-stimulated mobilization of hepatic glycogen stores. This can occur at birth or during the antepartum period. In the latter situation, fetuses might recover from stress and be delivered without difficulty. As newborns, such infants have depleted glycogen stores and are at risk for hypoglycemia.
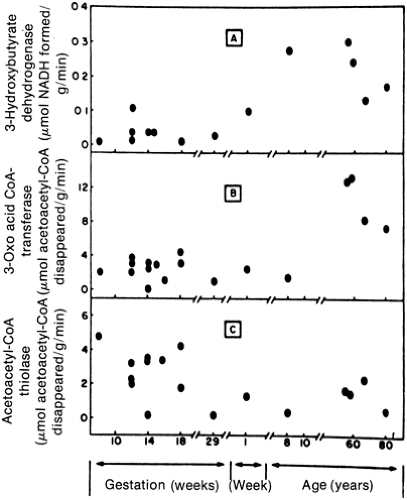 Figure 37-2 Enzymatic activity of the three key enzymes necessary for the oxidation of ketones (i.e., [A5]β-hydroxybutyrate and acetoacetate). The activities of these enzymes are present in substantial quantities in the human fetal brain during early gestation. (From Felig P, Lynch V. [Starvation in human pregnancy: hypoglycemia, hypoinsulinemia, and hyperketonemia.] Science 1970; 170:990-992, with permission.) |
Gluconeogenesis
For many years, it was believed that maternally derived glucose was the sole metabolic fuel for the fetus and that the fetus could not produce glucose. As indicated, the fetus can use other fuels such as the ketones and can, under special circumstances, mobilize hepatic glycogen. The fetus alsocan carry out gluconeogenesis to a limited degree, although it is likely that under normal circumstances it does not need to call on this function. Data from human abortuses have demonstrated that the four key gluconeogenic enzymes are present in fetal liver by 2 to 3 months of gestation (24,25). The activities of these
enzymes are believed to increase throughout gestation and the neonatal period. Thus, all appropriately grown newborns, including the very premature, probably have some degree of gluconeogenic capability. However, the growth-retarded neonate may have impaired gluconeogenic capability.
enzymes are believed to increase throughout gestation and the neonatal period. Thus, all appropriately grown newborns, including the very premature, probably have some degree of gluconeogenic capability. However, the growth-retarded neonate may have impaired gluconeogenic capability.
Endocrine Regulation
Insulin and glucagon, important hormones for regulating glucose, can be measured in fetal plasma as early as 12 weeks of gestation (26). Although plasma concentrations of these hormones are low, the relative content of these hormones in the fetal pancreas is quite high (27,28). These high concentrations may result from the limited ability of the fetal islets to secrete these hormones. Both premature and term infants have limited capacity to secrete these hormones in response to a glucose challenge in the newborn period. This indicates that the fetus also has limited secretory capability (Fig. 37-3) (28,29,30). Of note, amino acids have a greater effect in stimulating insulin and limiting glucagon secretion than glucose in the fetus (31,32).
Insulin may be more important for enhancing growth than for regulating metabolic fuels during fetal life. Insulin stimulates the growth of specific tissues (e.g., adipose, hepatic, connective, skeletal, cardiac muscle) (33,34). Excessive insulin secretion during fetal life resulting from such conditions as maternal diabetes causes the disproportionate growth of insulin-sensitive tissues, resulting in macrosomia (1,14,35,36). A lack of insulin, as in infants with transient neonatal diabetes mellitus, always is accompanied by fetal growth retardation.
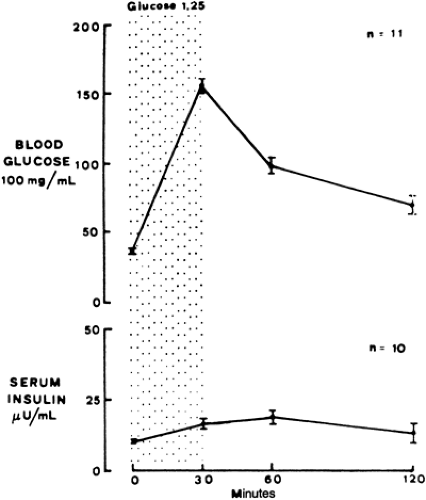 Figure 37-3 Insulin secretion after glucose challenge in premature infants. Whereas normal adults secrete insulin briskly in response to glucagon, premature infants in the neonatal period secrete insulin only sluggishly. (From Assan R, Buillet J. Pancreatic glucagon and glucagon-like material in tissues and plasma from human fetuses 6 to 26 weeks old. In: Jonxis JHP, Visser HKA, Troelstra JA, eds. Metabolic processes in the fetus and newborn infant, nutrition symposium. Baltimore: Williams & Wilkins, 1971: 210-212, with permission.) |
Glucagon or a critical glucagon/insulin ratio is important for inducing gluconeogenic enzymes. Glucagon stimulates the induction of gluconeogenic enzymes in vitro and in vivo (37,38). Fetal plasma glucagon concentrations increase progressively during fetal life, and this is associated with a concomitant increase in gluconeogenic enzyme activity. At birth, plasma glucagon concentrations surge, coinciding with the rapid postnatal increase in gluconeogenic activity (39). Insulin may modulate glucagon’s effect because it can inhibit gluconeogenic enzyme induction (37). Thus, a balance between these two hormones controls gluconeogenic enzyme induction during perinatal life.
Adrenergic mechanisms can stimulate hepatic glycogenol- ysis during fetal life, much as in the adult. As labor progresses, fetal sympathoadrenal activity increases, resulting in a considerable increase in circulating catecholamine levels (40,41). Cord clamping triggers an increase in glucagon secretion (42). As plasma glucose concentrations plummet with cord clamping, insulin secretion slowly decreases. These adjustments, particularly the remarkable increase in catecholamine secretion, stimulate glycogenolysis and gluconeogenesis in the neonate (Fig. 37-4).
Islet cell function remains unresponsive for several weeks of neonatal life in term infants. Newborn infants increase insulin and limit glucagon secretion sluggishly in response to glucose challenge. Limited data indicate that these responses become adult-like between the first and second weeks of life (43). This adaptation is critically important because the neonate, unlike the fetus, must
regulate glucose production and storage through feeding and fasting cycles. Little is known about the premature infant’s ability to regulate glucose in the neonatal period. The ability to modulate insulin and glucagon secretion probably develops as the neonatal period progresses.
regulate glucose production and storage through feeding and fasting cycles. Little is known about the premature infant’s ability to regulate glucose in the neonatal period. The ability to modulate insulin and glucagon secretion probably develops as the neonatal period progresses.
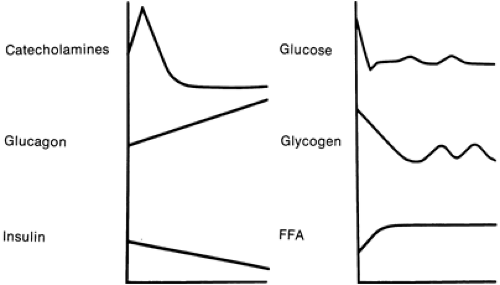 Figure 37-4 Levels of hormones and metabolic fuels change after birth. At birth, the counterregulatory hormones (i.e., catecholamines and glucagon) increase greatly, whereas insulin secretion decreases. Neonatal plasma glucose concentrations plummet as a result of cord clamping. The changes in counter regulatory hormones and insulin favor mobilization of glucose and fat and stimulate gluconeogenesis. These changes assure adequate neonatal glucose production. FAA, free fatty acids. (From Kalhan SC, Bier DM, Savin SM, et al. Estimation of glucose turnover and 13C recycling in the human newborn by simultaneous [1-13C]glucose and [6,6-1H2]glucose tracers. J Clin Endocrinol Metab 1980;50:456-460, with permission.) |
Glucose Transporters
Glucose transporters (Glut) are a family of structurally similar proteins encoded by a family of genes and expressed in a tissue-specific manner in most mammalian tissues (44). Several isoforms have been described in the human fetus and placenta. Because Gluts facilitate transfer of glucose from the maternal to fetal circulation and also uptake of glucose by most fetal and neonatal tissues, they are critically important for growth and development.
Glut1 is the dominant isoform in most fetal tissues and the placenta (45,46). Insulin, insulin-like growth factors, and other hormones and peptides regulate its activity and expression. The Gluts are developmentally regulated. The appearance of Glut isoforms varies in time of appearance and type (Glut2 in liver, Glut4 in muscle) during intrauterine and neonatal life (47). Glut1 and Glut3 are expressed in various tissues, including the placenta. Ambient glucose can affect both glucose transport activity and expression of Glut genes. Glut1 expression appears to increase with gestation. In placentas of women with diabetes, Glut3 decreases with gestation, suggesting an attempt by the placenta to limit glucose transport in the face of maternal hyperglycemia (48).
NEONATAL GLUCOSE REQUIREMENTS
The clinician makes a leap of faith in using the chemical or paper strip determination of plasma or blood glucose concentration to judge the adequacy of tissue glucose provision in the neonate. A normal plasma concentration is interpreted to mean that glucose supply to the brain and other organs is adequate for ongoing metabolic needs. To appreciate glucose requirements, glucose kinetics must be understood.
Glucose turnover represents the rate of production of glucose by the liver and other organs and the simultaneous use or uptake of glucose by the brain and other organs. Turnover is usually expressed as milligrams of glucose per kilogram of body weight per minute. Although stable isotope technology has allowed quantification of glucose turnover in neonates, this methodology cannot be directly applied for clinical purposes. Thus, the clinician must rely on the measurements of glucose concentrations, which are static, as representative of the dynamic rate of glucose production and use in a neonate. In general, plasma glucose concentrations roughly correlate with glucose turnover. Diminished plasma glucose concentrations suggest that glucose production is limited or that glucose use is increased (i.e., need is outstripping production). Elevated plasma glucose concentrations suggest that either production is excessive or, more likely, organ uptake and use are diminished. These are the dynamic physiologic conditions that define hypoglycemia and hyperglycemia.
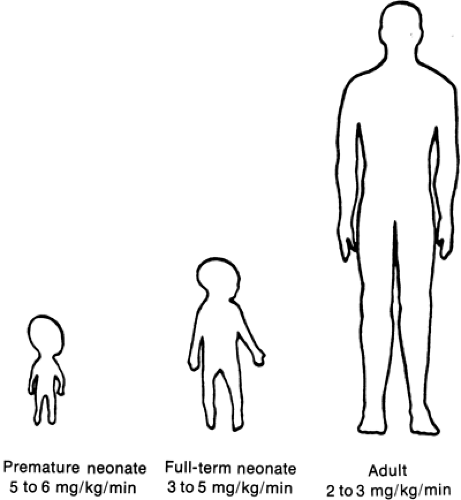 Figure 37-5 Glucose turnover in premature and term neonates and the adult. When related to body weight, glucose turnover is greatest in the premature infant and least in the adult. The increased turnover in neonates results in part from their relatively increased brain-to-body mass ratio. (From Kalhan SC, Bier DM, Savin SM, et al. Estimation of glucose turnover and 13C recycling in the human newborn by simultaneous [1-13C]glucose and [6,6-1H2]glucose tracers. J Clin Endocrinol Metab 1980;50:456-460, with permission.) |
In the neonate, glucose production correlates directly with brain and body mass, confirming the critical role of glucose as a metabolic fuel (49,50,51,52). This holds even in the most immature of premature infants (53). Glucose turnover for newborn infants, when related to body mass, significantly exceeds that of adults (Fig. 37-5). Premature infants have even greater turnover values than term neonates. This in part reflects the ratio of brain to body mass, which is greatest in premature infants and least in adults. These relations emphasize the importance of glucose as the primary fuel for the brain (Fig. 37-6).
Neonatal Hypoglycemia
A variety of blood and plasma glucose concentration values based on screening of neonates or clinical experience have been recommended as values defining hypoglycemia (54,55). All of these are somewhat arbitrary because they cannot be correlated directly with glucose use rate or severity of symptoms. Because plasma or blood glucose concentrations only roughly reflect glucose turnover, a plasma glucose concentration less than 40 mg/dL should be used to define hypoglycemia. When glucose turnover is sufficient to meet the needs of the organism, concentrations usually
exceed this value. It also is important to note that values somewhat less than 40 mg/dL still can be associated with adequate glucose provision.
exceed this value. It also is important to note that values somewhat less than 40 mg/dL still can be associated with adequate glucose provision.
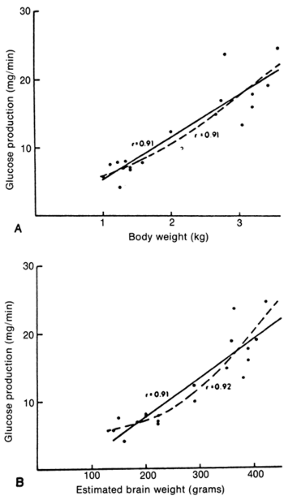 Figure 37-6 Linear and curvilinear regression analyses indicate strong relations between glucose turnover and body mass (A), and between glucose turnover and brain mass (B), in newborn human infants. (From Devaskar SU, Mueckler MM. The mammalian glucose transporters. Pediatr Res 1992;31:1-13, with permission. |
The chemical definition of hypoglycemia must take into account the methodology of glucose determination. Glucose concentration in whole blood is approximately 10% to 15% lower than that in plasma. Delay in determination after blood sampling may result in glucose oxidation by erythrocytes, causing falsely low values. Although the use of paper strip methods to estimate glucose concentrations quickly is acceptable, their results should be corroborated by true chemical determinations.
The clinical manifestations of inadequate glucose provision to the neonatal brain range from no symptoms to lethargy or mild tremors to frank convulsions (Table 37-1). The degree of glucose limitation necessary to cause brain damage is unknown. The lack of clearly defined data on this problem and the prevailing opinion concerning the potentially damaging effects of hypoglycemia mandate that infants at risk be monitored and that asymptomatic and symptomatic infants be appropriately treated.
TABLE 37-1 SYMPTOMS OF HYPOGLYCEMIA | |
|---|---|
|
All conditions associated with the development of hypoglycemia in the neonate result from one or a combination of two basic mechanisms: inadequate production or excessive tissue use. Inadequate glucose production results from a lack of glycogen stores, an inability to synthesize glucose, or both (Fig. 37-7). Excessive tissue use results from increased insulin secretion. Table 37-2 categorizes infants at risk for hypoglycemia in relation to these basic mechanisms.
Inadequate Glucose Production as a Result of Limited Glycogen Stores
Premature Infants
As indicated, the third trimester of pregnancy is an important period for hepatic glycogen deposition. An infant delivered prematurely without the benefit of part of or the entire third trimester will have limited hepatic glycogen stores. The greater the degree of prematurity, the less glycogen will be present. SGA premature infants are at extremely
high risk for development of hypoglycemia because available nutrients during intrauterine life are channeled toward growth, with little set aside for glycogen storage. For this reason, SGA premature infants have extremely limited glycogen stores (55,56,57).
high risk for development of hypoglycemia because available nutrients during intrauterine life are channeled toward growth, with little set aside for glycogen storage. For this reason, SGA premature infants have extremely limited glycogen stores (55,56,57).
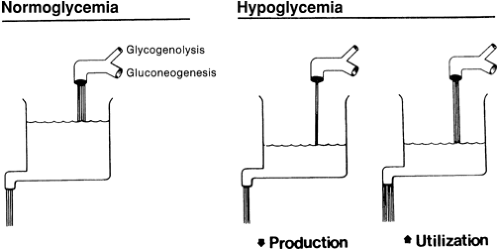 Figure 37-7 The rates of glucose production and utilization are represented by the faucet and drain of the sink. The level in the sink is equivalent to plasma or blood glucose concentrations. If production from glycogenolysis and gluconeogenesis is adequate, and use is not excessive, normoglycemia exists and the plasma or blood glucose concentration (i.e., the level in the sink) is normal. Hypoglycemia develops if production is inadequate to meet body needs or if use outstrips production. This results in decreased glucose concentrations (i.e., diminished level in sink). (From Kalhan SC, Bier DM, Savin SM, et al. Estimation of glucose turnover and 13C recycling in the human newborn by simultaneous [1-13C]glucose and [6,6-1H2]glucose tracers. J Clin Endocrinol Metab 1980; 50:456-460, with permission.) |
TABLE 37-2 INFANTS AT RISK FOR HYPOGLYCEMIAa | ||
|---|---|---|
|
Infants Who Have Suffered Perinatal Disease
Infants who are stressed in utero are at increased risk for development of hypoglycemia as neonates. Hypoxia, acidosis, and alterations in fetal blood pressure and flow can stimulate catecholamine secretion in utero, which in turn will mobilize hepatic glycogen stores. In addition, hypoxia increases the rate of anaerobic glycolysis, thereby accelerating glucose use. These events deplete fetal glycogen stores and place the infant at risk for hypoglycemia after delivery.
Glycogen Storage Disease
Intrinsic defects in glycogen synthesis, storage, or breakdown result in complex metabolic problems (see Chapter 41). Several types of glycogen storage disease (e.g., Ia, I, VI, 0) include hypoglycemia as one of many associated complications (54,58).
Inadequate Glucose Production as a Result of Limited Gluconeogenesis
Small-for-Gestational-Age Infants
Full-term and premature SGA neonates are at great risk for development of hypoglycemia as a result of inadequate hepatic glycogen stores. With treatment, the hypoglycemia is usually short lived. Approximately 1% of SGA infants in whom hypoglycemia develops have a prolonged course requiring intravenous therapy for days. A delay in the induction of gluconeogenic capability probably is responsible for this prolonged hypoglycemia. These SGA neonates have elevated plasma concentrations of gluconeogenic precursors, suggesting an inability to convert exogenous gluconeogenic precursors, such as alanine, to glucose (59,60).
In animal models of intrauterine growth retardation, the induction of one gluconeogenic enzyme, phosphoenolpyruvate carboxykinase, is delayed (61,62). This occurs despite appropriate increases in glucagon and decreases in insulin; these relations should favor enzyme induction. Why some SGA infants fail to induce gluconeogenic capability is unclear. Corticosteroid therapy can be used to treat prolonged hypoglycemia. The success of this practice probably results in part from the ability of corticosteroids to induce hepatic gluconeogenic enzymes.
Many SGA infants have heightened metabolic requirements during the neonatal period. The mechanisms for this are not understood, but they may represent an attempt to compensate for the preceding intrauterine deprivation (63). This may explain the increased glucose requirements demonstrated by some hypoglycemic SGA infants.
Congenital Absence of Gluconeogenic Capability
Unlike SGA infants in whom gluconeogenic capability eventually develops, a few infants have been reported to have permanent congenital lack of gluconeogenic enzymes (64). A single infant who was unable to secrete glucagon also has been reported (65).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


