Chapter 491 Brain Tumors in Childhood
Etiology
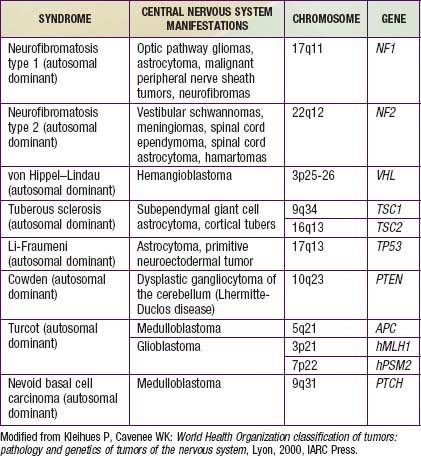
The etiology of pediatric brain tumors is not well defined. A male predominance is noted in the incidence of medulloblastoma and ependymoma. Familial and hereditary syndromes associated with increased incidence of brain tumors account for approximately 5% of cases (Table 491-1). Cranial exposure to ionizing radiation also is associated with a higher incidence of brain tumors. There are sporadic reports of brain tumors within families without evidence of a heritable syndrome. The molecular events associated with tumorigenesis of pediatric brain tumors are not known.
Pathogenesis
More than 100 histologic categories and subtypes of primary brain tumors are described in the World Health Organization (WHO) classification of tumors of the CNS. In children 0-14 yr, the most common tumors are pilocytic astrocytomas (PAs) and medulloblastoma/primitive neuroectodermal tumors (PNETs). In adolescents (15-19 yr), the most common tumors are pituitary tumors and PAs (Fig. 491-1).
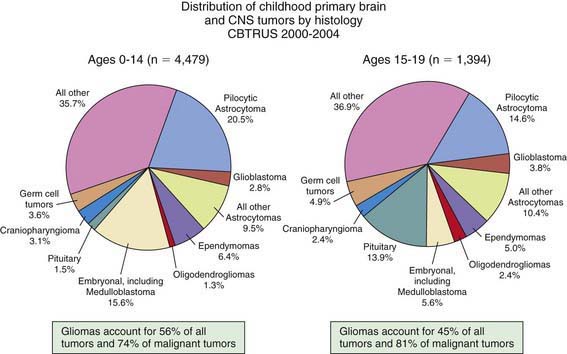
Figure 491-1 Distribution of childhood primary brain and CNS tumors by histology.
(From Central Brain Tumor Registry of the United States [CBTRUS]: CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2004-2006: February 2010 (PDF file). www.cbtrus.org/2010-NPCR-SEER/CBTRUS-WEBREPORT-Final-3-2-10.pdf. Accessed March 19, 2011.)
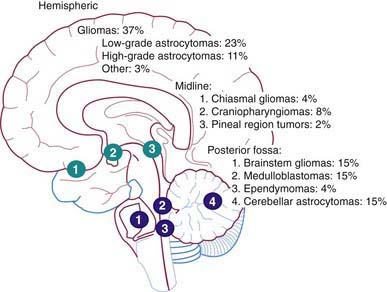
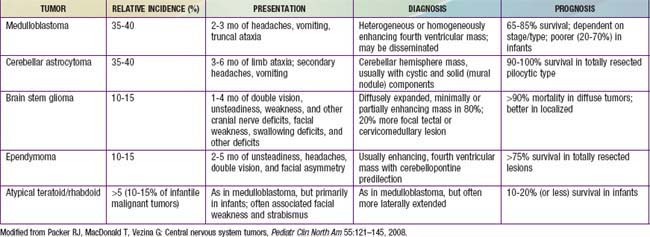
The Childhood Brain Tumor Consortium reported a slight predominance of infratentorial tumor location (43.2%), followed by the supratentorial location (40.9%), spinal cord (4.9%), and multiple sites (11%) (Fig. 491-2, Table 491-2). There are age-related differences in primary location of tumor. During the first year of life, supratentorial tumors predominate and include, most commonly, choroid plexus complex tumors and teratomas. In children 1-10 yr of age, infratentorial tumors predominate, owing to the high incidence of juvenile pilocytic astrocytoma and medulloblastoma. After 10 yr of age, supratentorial tumors again predominate, with diffuse astrocytomas most common. Tumors of the optic pathway and hypothalamus region, the brainstem, and the pineal-midbrain region are more common in children and adolescents than in adults.
Specific Tumors
Astrocytomas
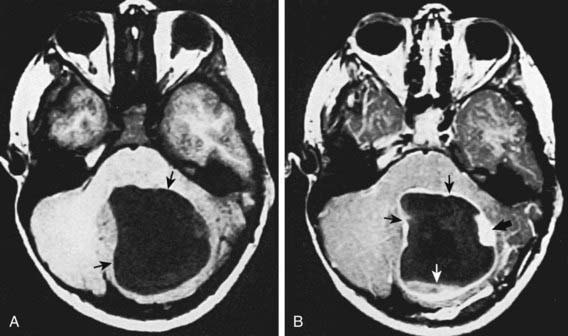
Low-grade astrocytomas (LGAs), the predominant group of astrocytomas in childhood, are characterized by an indolent clinical course. PA is the most common astrocytoma in children, accounting for about 20% of all brain tumors (Fig. 491-3). On the basis of clinicopathologic features using the WHO Classification System, PA is classified as a WHO grade I tumor. Although PA can occur anywhere in the CNS, the classic site is the cerebellum. Other common sites include the hypothalamic/third ventricular region and the optic nerve and chiasmal region. The classic but not exclusive neuroradiologic finding in PA is the presence of a contrast medium–enhancing nodule within the wall of a cystic mass (see Fig. 491-3). The microscopic findings include the biphasic appearance of bundles of compact fibrillary tissue interspersed with loose microcystic, spongy areas. The presence of Rosenthal fibers, which are condensed masses of glial filaments occurring in the compact areas, helps establish the diagnosis. PA has a low metastatic potential and is rarely invasive. A small proportion of these tumors can progress and develop leptomeningeal spread, particularly when they occur in the optic path region. A PA very rarely undergoes malignant transformation to a more aggressive tumor. A PA of the optic nerve and chiasmal region is a relatively common finding in patients with neurofibromatosis type 1 (15% incidence). Unlike in diffuse fibrillary astrocytomas, there are no characteristic cytogenetic abnormalities in PA nor are there any known molecular abnormalities. Other tumors occurring in the pediatric age group with clinicopathologic characteristics similar to those of PA include pleomorphic xanthoastrocytoma, desmoplastic cerebral astrocytoma of infancy, and subependymal giant cell astrocytoma.
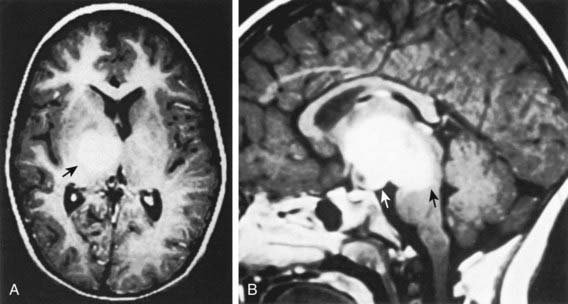
The second most common astrocytoma is fibrillary infiltrating astrocytoma, which consists of a group of tumors characterized by a pattern of diffuse infiltration of tumor cells among normal neural tissue and potential for anaplastic progression. On the basis of their clinicopathologic characteristics, they are grouped as low-grade astrocytomas (WHO grade II), malignant astrocytomas (anaplastic astrocytoma; WHO grade III), and glioblastoma multiforme (GBM; WHO grade IV). Of this group, the fibrillary LGA is the second most common astrocytoma in children, accounting for 15% of brain tumors. Histologically, these low-grade tumors demonstrate greater cellularity than normal brain parenchyma, with few mitotic figures, nuclear pleomorphism, and microcysts. The characteristic MRI finding is a lack of enhancement after contrast agent infusion (Fig. 491-4). Molecular genetic abnormalities found among low-grade diffuse infiltrating astrocytomas include mutations of p53 and overexpression of platelet-derived growth factor α-chain and platelet-derived growth factor receptor-α. Fibrillary infiltrating astrocytoma has the potential to evolve into malignant astrocytoma, a development that is associated with cumulative acquisition of multiple molecular abnormalities.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


