Fig. 1
Microscopic view of an aneurysmal bone cyst. Solid area consisting of reactive bone formation (*) with prominent giant cells (arrows) in a background of spindle cells with vascular proliferation. There is no notable mitotic activity identified and no cytological atypia. (a) 100× magnification. (b) 200× magnification (Courtesy of Joshua M. Abzug, MD)
Pathoanatomy and Applied Anatomy Relating to Aneurysmal Bone Cyst
The majority of cysts that develop in the hand affect the metacarpals and phalanges. Due to the rarity of these lesions, aneurysmal bone cysts are often misdiagnosed as more common tumors such as giant cell tumors, benign cartilage tumors, or interosseous ganglion (Mankin et al. 1995). Progression of aneurysmal bone cysts has been described in four phases. The initial phase is characterized by osteolysis of the marginal aspect of the bone with periosteal elevation. The growth phase is indicated by progressive destruction of the bone leading to poor demarcation of the lesion. The stabilization phase is identified by the classic appearance of the cyst with a well-defined bony shell and osseous septations. Lastly, the healing phase is observed when advancing ossification of the lesion is apparent (Dabaska and Buraczewski 1969; Rapp et al. 2012).
Initially, ABCs were thought to develop in response to an intraosseous hemorrhage, leading to formation of a cyst (Lichtenstein 1950; Ratcliffe and Grimer 1993). However, recent genetic and histopathologic studies have suggested that aneurysmal bone cysts are likely to represent true neoplasms undergoing tumorigenesis rather than reactive lesions (Oliveira et al. 2004; Ye et al. 2010; Fig. 1).
Assessment of Aneurysmal Bone Cyst
Signs and Symptoms of Aneurysmal Bone Cyst
Patients with aneurysmal bone cysts commonly present with localized pain and swelling or following a pathological fracture.
Aneurysmal Bone Cyst Imaging and Other Diagnostic Studies
Plain radiographs will demonstrate lytic metaphyseal lesions with a classic “eggshell” sclerotic border. These lesions are well circumscribed, and radiographs may also show a “soap bubble” appearance due to the remaining trabeculae supporting the bone. The cortex is typically thin but intact (Copley and Dormans 1996). If operative intervention is pursued, computed tomography may be utilized preoperatively to define the boundaries of the lesion (Fig. 2).
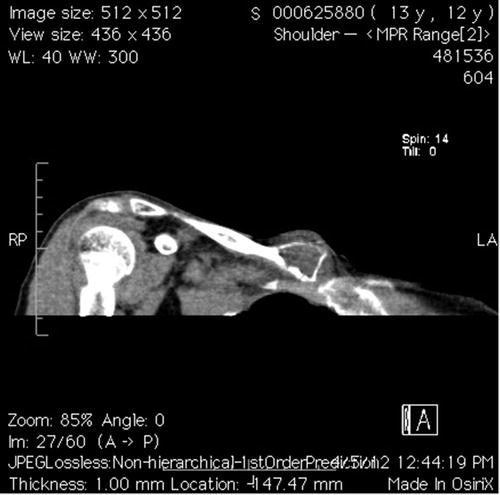
Fig. 2
Coronal CT scan showing an aneurysmal bone cyst of the medial clavicle. Advanced imaging aided in making the correct diagnosis and assessing the extent of the lesion (Courtesy of Joshua M. Abzug, MD)
Magnetic resonance imaging with contrast commonly demonstrates osseous internal septations, often containing fluid-fluid levels (Fig. 3). Although this finding is highly suggestive of an aneurysmal bone cyst, the surgeon should not rule out telangiectatic osteosarcoma, giant cell tumor, a secondary aneurysmal bone cyst, or a fracture through a simple cyst as all of these may have fluid-fluid levels within them as well (Rapp et al. 2012). When diagnosing aneurysmal bone cysts, it has been reported that MRI combined with conventional radiography allows for the greatest sensitivity, specificity, and positive predictive value (Mahnken et al. 2003).
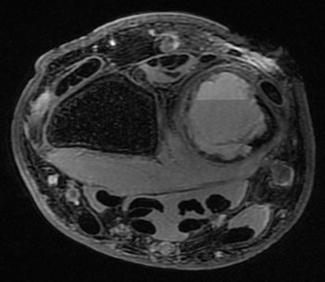
Fig. 3
Axial MRI of an aneurysmal bone cyst in the distal ulna. Note the fluid-fluid level (Courtesy of Joshua M. Abzug, MD)
Injuries Associated with Aneurysmal Bone Cyst
Pathological fractures can occur in association with aneurysmal bone cysts in the setting of minor trauma. Additionally, lesions that cross the growth plate will like result in a partial or complete growth arrest.
Aneurysmal Bone Cyst Treatment Options
Nonoperative Management of Aneurysmal Bone Cyst
Indications/Contraindications
Observation may be considered in asymptomatic patients with an aneurysmal bone cyst.
Techniques
Observation is an active process, requiring serial physical examinations and radiographs every 4–6 months to evaluate for symptoms and radiologic evidence of progression. If the diagnosis is unclear, a biopsy should be performed to ensure accurate diagnosis.
Operative Treatment of Aneurysmal Bone Cyst
Indications/Contraindications
Operative management of aneurysmal bone cysts should be considered if the cyst is painful, growing, or at a site that may lead to a fracture.
Surgical Procedure
Aneurysmal bone cysts are most commonly treated with excision, curettage, and bone grafting. Cryosurgery has been successfully utilized to treat recurrent lesions in the proximal phalanx, while wide excision has been successful in treating metacarpal cysts (Borrelli and McCormack 1994; Burkhalter et al. 1978; Frassica et al. 1988; Fuhs and Herndon 1979; Marcove et al. 1995). Amputation may be required when treating large aneurysmal bone cysts of the distal phalanx (Fuhs and Herndon 1979).
Preoperative Planning
Bone grafting is typically performed following the curettage, and therefore, a source for the graft should be considered. Additionally, the specimen is often sent for frozen section, so the surgeon should discuss this with the pathologist prior to proceeding to the operating room. Intraoperative imaging should also be available via radiographs or fluoroscopy.
Positioning
The patient is typically in the supine position with the arm extended on a radiolucent hand table. A modified beach chair position may be utilized for lesions of the proximal humerus.
Surgical Approach(es)
A direct approach to the lesion is performed, as a small incision is all that is needed to curettage and graft an ABC.
Technique
Via a 2–3 cm incision, blunt dissection is carried down to bone. A small corticotomy is made with either a drill bit or a curette. Subsequently, blood should be visible, consistent with the diagnosis of an ABC. Curettes are then utilized to curettage the cyst cavity. It is important to “break through” to the medullary canal as this is theorized to improve the healing of the cyst. Following a thorough curettage, the defect is grafted (Fig. 4).
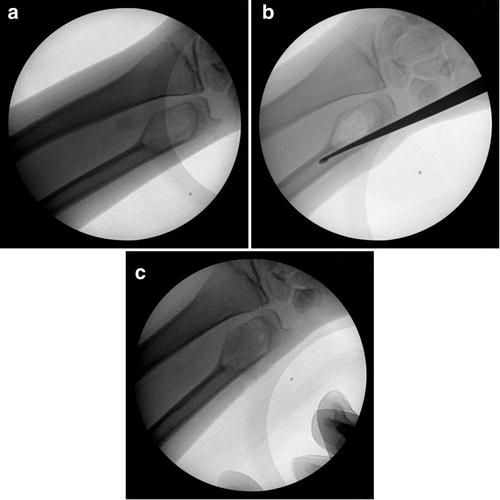
Fig. 4
(a) Aneurysmal bone cyst of the distal ulna. (b) Curettage of the cyst cavity followed by “breaking through” to connect the cyst cavity with the medullary canal. (c) Grafting of the cyst following curettage (Courtesy of Joshua M. Abzug, MD)
Treatment-Specific Outcomes
Preferred Treatment
The majority of ABCs are treated surgically with curettage and grafting. A direct approach via a 2–3 cm incision is utilized. Subsequently, a corticotomy is made with the curette as the cortex is quite thin. The cyst is then curettaged thoroughly and a connection is made with the medullary canal. Injectable synthetic bone graft is utilized to fill the remaining defect (Fig. 4).
Surgical Pitfalls and Prevention
It is important to ensure that the adjacent physis is not damaged and that an intraoperative fracture does not occur. This can be done by judicious use of fluoroscopy during the curettage portion of the procedure.
Management of Complications
Recurrence of the cyst or failure of the cyst to heal is treated by repeating the curettage and grafting procedure.
Bizarre Parosteal Osteochondromatous Proliferation
Bizarre parosteal osteochondromatous proliferation (BPOP, Nora’s lesion) is a benign, reactive proliferation of bone and cartilage that is often associated with a traumatic injury to the hand (Orui et al. 2002; Payne and Merrell 2010). These lesions arise from cortical bone and are not continuous with the medullary cavity. Plain radiographs typically demonstrate a well-defined calcified mass arising from the surface of the bone (Michelsen et al. 2004; Orui et al. 2002). Histology demonstrates a lesion with a cartilage cap comprised of bizarre-appearing chondrocytes and hypercellularity (Payne and Merrell 2010). Due to the aggressive appearance on radiographs and histology, BPOP may initially be mistaken for a parosteal osteogenic sarcoma. Surgical excision is the treatment of choice for these lesions despite the high risk for local recurrence. In order to decrease the risk of recurrence, Michelsen et al. have suggested excising the underlying periosteum as well (Michelsen et al. 2004). Wide or radical excision is generally unwarranted due to the benign clinical course.
Chondromyxoid Fibroma
Chondromyxoid fibroma is a rare benign cartilaginous tumor that accounts for less than 1 % of all bone tumors. This lesion typically presents in the second or third decade of life, with males affected twice as often as females (Nalbantoglu et al. 2005).
Pathoanatomy and Applied Anatomy Relating to Chondromyxoid Fibroma
Chondromyxoid fibromas are usually located in an eccentric metaphyseal location, but in the hand, they are more commonly found centrally (Payne and Merrell 2010; Nalbantoglu et al. 2005; Strauch and Kleinman 1996). Cortical expansion along with the presence of a sclerotic rim is typically present. Additionally, pseudotrabeculation may be seen. Histologically, chondromyxoid fibromas are derived from cartilage and are comprised of varying amounts of chondroid, fibrous, and myxoid tissue. Due to the aggressive histological appearance, chondromyxoid fibroma may be confused with chondrosarcoma.
Assessment of Chondromyxoid Fibroma
Signs and Symptoms of Chondromyxoid Fibroma
Approximately 70 % of patients complain of symptoms at the time of diagnosis (Wu et al. 1998). Patients commonly present with mild chronic pain or swelling that may have been present for years.
Chondromyxoid Fibroma Imaging and Other Diagnostic Studies
Chondromyxoid fibromas appear on radiographs as well-defined, elongated, radiolucent lesions in the metaphysis of long bones. These lesions typically exhibit sclerotic borders with mild cortical expansion and are eccentrically located. However, lesions of the bones of the hands are commonly more centrally located. Chondromyxoid fibromas are generally between 1 and 10 cm in diameter and may extend into the diaphysis or epiphysis (Marin et al. 1997; Merine et al. 1989).
Injuries Associated with Chondromyxoid Fibroma
A pathological fracture associated with a chondromyxoid fibroma occurs in approximately 5 % of patients (Giudici et al. 1993).
Chondromyxoid Fibroma Treatment Options
Nonoperative Management of Chondromyxoid Fibroma
Indications/Contraindications
There is a limited role for nonoperative management of chondromyxoid fibromas as they are typically symptomatic and have an appearance on radiographs that can be worrisome.
Techniques
Nonsteroidal anti-inflammatory medications or other analgesics may be used to control the chronic pain produced by chondromyxoid fibromas while one is awaiting surgical intervention.
Outcomes
No specific outcomes exist regarding the nonoperative management of chondromyxoid fibromas in the pediatric upper extremity.
Operative Treatment of Chondromyxoid Fibroma
Indications/Contraindications
As most lesions are painful and may possess worrisome features on radiographs, surgery is usually performed.
Surgical Procedure
Preoperative Planning
The pathologist needs to be aware that the procedure is being performed and that a frozen section is being sent. Additionally, graft material needs to be readily available.
Positioning
Patients are placed supine on the operating room table and a radiolucent hand table is utilized.
Surgical Approach(es)
A direct approach to the tumor is utilized following the principles of tumor surgery including the utilization of a longitudinal incision.
Technique
Curettage and grafting are performed under fluoroscopic guidance utilizing curettes. It is essential to ensure that the physis is not damaged; however, these lesions have the potential to cross the physis.
Treatment-Specific Outcomes
Preferred Treatment
Curettage and grafting are performed with the utilization of synthetic graft materials. For lesions in the digits, a lateral approach is preferred to limit scarring potential of the extensor mechanism.
Surgical Pitfalls and Prevention
It is essential to avoid damaging the physis during the curettage of the lesion. Additionally, one must be careful to avoid causing an intraoperative fracture, as the cortex may be thinned. Both of these can be avoided by using fluoroscopy judiciously during the procedure.
Management of Complications
Recurrence of the lesion is treated with repeat curettage and grafting.
Enchondroma
Enchondroma is a benign intramedullary cartilaginous neoplasm. It is the most common primary bone tumor of the hand, accounting for approximately 90 % of bone tumors in the hand. Furthermore, approximately 35 % of all enchondromas develop in the hand (Bauer et al. 1988). Enchondromas typically occur in younger patients with more than 50 % of patients between 11 and 30 years of age (Nurboja et al. 2006).
Pathoanatomy and Applied Anatomy Relating to Enchondroma
An enchondroma results due to dysplasia of the central portion of the physis leading to failure of normal endochondral ossification (Dietz et al. 2007). Chondroblasts normally located in the physis may escape into the metaphysis, where they proliferate. Thus, the most observed site of enchondroma formation is within the medullary cavity of the diaphysis or metaphysis, most commonly in the hand. These lesions are most often at the central metaphysis of the proximal phalanx, followed by the metacarpal and middle phalanx. Other locations in the upper extremity that have been reported to have enchondromas include the scaphoid, lunate, capitate, and proximal humerus (Dietz et al. 2007).
Assessment of Enchondroma
Signs and Symptoms of Enchondroma
The majority of patients with a solitary enchondroma present due to painless or painful swelling or following a pathological fracture from minor trauma. Often, these lesions are diagnosed as an incidental finding on plain radiographs. If the enchondroma is symptomatic, patients may note widening of the bone/finger, angular deformity, or a limb-length discrepancy.
Enchondroma Imaging and Other Diagnostic Studies
Plain radiographs are the imaging modality of choice for visualization of the vast majority of enchondromas. Typically a well-circumscribed lytic lesion is present in the metaphysis or diaphysis that may be lobulated. Expansion and thinning of the surrounding cortex may be evident as well. CT scans and MRI are generally not necessary.
Injuries Associated with Enchondroma
The most common injury associated with a solitary enchondroma is a pathological fracture.
Enchondroma Treatment Options
Nonoperative Management of Enchondroma
Indications/Contraindications
Observation is the treatment of choice for small asymptomatic lesions with a typical radiographic appearance. Large or symptomatic enchondromas, however, should be managed by biopsy and curettage with grafting, as an impending pathological fracture may occur. Lesions leading to pathological fractures may be treated acutely or, more commonly, after the fracture has healed. However, there are no studies demonstrating improved clinical outcomes if treatment is performed after the pathological fracture has healed.
Techniques
Serial radiographs every 6–12 months are indicated during observation to follow the course of the enchondroma and to ensure that there has been no significant change in the size of the tumor.
Outcomes
Solitary enchondromas are generally self-limited, but a certain percentage of them will continue to grow. Delayed union or nonunion occurs in approximately 15 % of children who have had one or more pathological fractures (Dietz et al. 2007).
Operative Treatment of Enchondroma
Indications/Contraindications
Indications for surgical management of an enchondroma include lesions that demonstrate any significant change in size or appearance on serial radiographs, radiographs that are suspicious for the presence of a low-grade chondrosarcoma, and large lesions that are at risk for an impending pathological fracture, particularly in the digits.
Surgical Procedure
Intralesional curettage with bone grafting is the mainstay of surgical treatment of enchondromas. Immobilization prior to curettage and bone grafting may be warranted if there is a pathological fracture that has occurred. This can be helpful to ensure that the bone graft does not leak out to surrounding tissues during the curettage and grafting procedure.
Preoperative Planning
Preoperatively, the radiographic extent and severity of the tumor should be assessed. The surgeon should determine the approach based on the anatomic location of the lesion. Bone grafting is almost universally performed, and therefore, a source for autograft should be considered or one needs to make sure allograft or synthetic graft is available. There may be a need for frozen section depending on whether or not there is concern for a low-grade chondrosarcoma, and therefore, the surgeon should ensure that the pathologist is aware and available prior to proceeding to the operating room. Intraoperative imaging should also be available via plain radiographs and/or fluoroscopy.
Positioning
The procedure is usually performed in the supine position with the arm extended on a radiolucent hand table. A modified beach chair position may be necessary for lesions of the proximal humerus.
Surgical Approach(es)
Lesions of the phalanx can be approached via a dorsal or lateral approach, while metacarpal and carpal lesions are usually approached utilizing a dorsal incision. Distal radius lesions can be approached dorsally at the level of Lister’s tubercle with a limited incision needed.
Technique
Prior to performing a wide exposure, an initial biopsy should be performed through a limited exposure so that a preliminary diagnosis can be confirmed on frozen section. Once a benign process is confirmed, a thorough curettage of the enchondroma should be performed utilizing a combination of curved and straight curettes. Intraoperative imaging should be utilized to ensure that the entire lesion is curettaged without damaging the physis. Subsequently, the graft material should be placed into the lesion utilizing the corticotomy site. Options for graft material include autograft, easily obtained from the distal radius, allograft, and synthetic substitutes such as injectable calcium phosphate/sulfate bone cement (Lin et al. 2013; Yasuda et al. 2006). It is not well established whether grafting the defect reduces the risk of a subsequent fracture; however, it does provide short-term structural support following the curettage.
Treatment-Specific Outcomes
Controversy exists regarding whether or not filling the defect is necessary. Tordai et al. treated 46 enchondromas of the hand by simple curettage without bone grafting and reported 82 % of the defects healed and only 16 % were left with small bony defects. Only one patient had a pronounced recurrence requiring reoperation (Tordai et al. 1990). The recurrence rate of solitary enchondromas following intralesional curettage and bone grafting is less than 5 % (Bauer et al. 1995).
Preferred Treatment
A lateral approach to address phalangeal lesions is performed rather than a dorsal approach in order to reduce scarring of the extensor mechanism or contamination with the graft material. Following curettage, the cystic cavity is filled with an osteoinductive/osteoconductive synthetic product.
Surgical Pitfalls and Prevention
It is important to ensure that the physis is not damaged during the curettage of the tumor. Therefore, judicious use of fluoroscopy should be performed while the curettage is occurring. It is also important to ensure that the curettage is not too aggressive to cause a pathological fracture, as the cortices around the lesion may be quite thin. However, one should try to remove as much of the enchondroma as possible (Table 1).
Table 1
Enchondroma: Potential pitfalls and preventions
Potential pitfall | Pearls for prevention |
|---|---|
Damage to the physis | Judicious use of fluoroscopy |
Pathological fracture | Avoid aggressive curettage |
Management of Complications
The most common complication following treatment of an enchondroma is recurrence of the tumor. Treatment consists of repeat curettage and grafting. It is essential that any recurrence has a repeat biopsy sent to ensure that the original diagnosis was correct. Other potential complications include infection, persistent pain, stiffness, and subsequent fracture. Stiffness may require tenolysis of the extensor mechanism if a dorsal incision to a finger was utilized, especially if graft material inadvertently escapes and became interposed between the bone and the tendon.
Giant Cell Tumor of Bone
Giant cell tumor (GCT) of bone is an uncommon tumor that accounts for approximately 5 % of bone tumors (Athanasian et al. 1997). Although classified as benign based on histology, GCTs of bone may be locally aggressive, metastasize, and ultimately be fatal.
Pathoanatomy and Applied Anatomy Relating to Giant Cell Tumor of Bone
GCTs of bone typically affect the epiphyses of long bones, with the distal radius being the most common location in the upper extremity (Harness and Mankin 2004). Tumors can also be present in the metacarpals, phalanges, and carpal bones, but occurrence in these locations is exceedingly rare (Athanasian et al. 1997; FitzPatrick and Bullough 1977; Lane et al. 1994).
Assessment of Giant Cell Tumor of Bone
Signs and Symptoms of Giant Cell Tumor of Bone
The most common initial presenting symptoms of patients with GCT of bone include pain, swelling, and impairment of joint mobility.
Giant Cell Tumor of Bone Imaging and Other Diagnostic Studies
Giant cell tumors of bone typically require a combination of several imaging modalities to correctly determine the extent of disease. Plain radiographs demonstrate an eccentrically located, lytic, expansile lesion with no matrix and indistinct borders involving the epiphysis and adjacent metaphysis. CT scans provide a more accurate representation of cortical thinning and penetration. Assessment of bone mineralization can also be performed via the CT scan. MRI may be utilized in order to evaluate the surrounding soft tissue and neurovascular structures, as well as extension into adjacent joint spaces (Kwon et al. 2007). In the case of multicentric disease, a radionuclide bone scan may be warranted.
Giant Cell Tumor of Bone Classification
Campanacci et al. proposed a classification scheme describing the GCTs of bone as stage 1, 2, or 3. Stage 1 lesions are confined entirely within the bone and have limited cortical thinning, stage 2 lesions exhibit more extensive cortical damage and expansion, and stage 3 lesions have a marked amount of cortical damage and have to spread beyond the bone to produce a soft tissue mass (Campanacci et al. 1987).
Giant Cell Tumor of Bone Treatment Options
Nonoperative Management of Giant Cell Tumor of Bone
Indications/Contraindications
All suspected giant cell tumors of bone should be treated surgically due to their local aggressive behavior and ability to metastasize.
Operative Treatment of Giant Cell Tumor of Bone
Indications/Contraindications
Surgical intervention is the treatment of choice for resectable GCT of bone. Intralesional curettage, marginal excision, wide local excision, and en bloc resection have all been used, depending on the location and size of the tumor.
Surgical Procedure
Preoperative Planning
Preoperatively, the radiographic extent and severity of the tumor should be assessed. Bone grafting may be necessary, and therefore, a source for the graft should be considered. The surgeon should discuss the need for a frozen section with the pathologist prior to proceeding to the operating room. Intraoperative imaging should also be available via radiographs or fluoroscopy.
Positioning
The procedure is performed in the supine position with the arm extended on a radiolucent hand table.
Surgical Approach(es)
Due to the tendency of GCTs of bone to seed soft tissues, incisional biopsy should be carefully performed via a well-planned approach with soft tissue protection so that the risk of local soft tissue recurrence is decreased (Athanasian et al. 1997). Lesions of the phalanx can be approached via the dorsal or lateral approach, while metacarpal and carpal lesions are best approached dorsally. Distal radius lesions can be approached dorsally or volarly, depending on surgeon preference.
Technique
Surgical excision is the mainstay of treatment for giant cell tumors of bone, but the technique varies based on location of the tumor. Lesions of the proximal carpal have been treated with either curettage or proximal row carpectomy (Athanasian et al. 1997; Lane et al. 1994). Distal carpal lesions may require wide excision (Lane et al. 1994). Lesions of the distal radius may be treated by marginal excision or curettage and grafting, in an attempt to preserve the articular surface.
Treatment-Specific Outcomes
In a systematic review of treated GCTs of the small bones of the hands and feet by Oliveira et al., there was a 72 % recurrence rate in patients treated with isolated curettage, compared to 15 % in patients treated by resection and 10 % treated by amputation (Oliveira et al. 2013).
Preferred Treatment
GCTs of bone are treated with marginal excision as opposed to curettage and grafting, due to their high recurrence rate. However, the articular surface should try to be preserved during the process.
Surgical Pitfalls and Prevention
It is imperative to excise the entire tumor, as local recurrence can be aggressive and even lead to metastases.
Management of Complications
Recurrent lesions should be treated with a repeat excision of the tumor utilizing a wider margin.
Maffucci’s Syndrome
Maffucci’s syndrome is a rare, congenital enchondromatosis similar to Ollier’s disease; however, patients with Maffucci’s syndrome additionally exhibit multiple hemangiomas. The hands are affected in the majority of cases of Maffucci’s syndrome. Radiographically, Maffucci’s syndrome appears identical to Ollier’s disease, except for the presence of phleboliths, which correspond to the hemangiomas. Deformity of the hands may occur, and patients with this syndrome are at high risk for the development of both bone and soft tissue sarcomas (Jacobs et al. 2010).
Multiple Enchondromatosis (Ollier’s Disease)
Multiple enchondromatosis (Ollier’s disease) is a rare, nonhereditary condition in which multiple enchondromas occur, predominantly in a unilateral distribution (Fig. 5) (Fang et al. 2009). The disease typically affects the metaphysis and diaphysis of long bones or short tubular bones of the hands and feet (Bükte et al. 2005). Ollier’s disease has an incidence of approximately 1:100,000, with females more commonly affected (Fang et al. 2009; Van Loon and Lammens 2008).
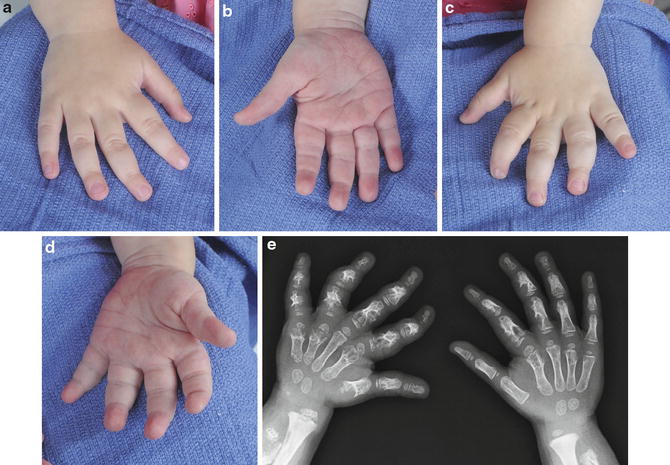
Fig. 5
Four-year-old female with multiple enchondromatosis or Ollier’s disease (Courtesy of Shriners Hospital for Children). (a) Dorsum of the right hand. (b) Palm of the right hand. (c) Dorsum of the left hand. (d) Palm of the left hand. (e) Bilateral hand x-rays with extensive bony involvement
Evidence of Ollier’s disease generally presents prior to puberty, as the lesions become more apparent with progressive skeletal growth (Bükte et al. 2005). Therefore, it is important to note that growth of an enchondroma after skeletal maturity may indicate malignant transformation. Approximately 30 % of patients with Ollier’s disease will develop a malignant bone neoplasm, specifically chondrosarcoma or osteosarcoma. Thus, patients with Ollier’s disease should undergo regularly scheduled serial physical and radiologic examinations. Other sequelae of Ollier’s disease can include skeletal deformities and/or a limb-length discrepancy (Van Loon and Lammens 2008). Progressive nonmalignant deformity can be treated with a diaphysectomy and fibula strut grafting (Fatti and Mosher 1986). Partial resection of the cortical bone with curettage of the tumor (corticoplasty) has also been shown to be successful in treating Ollier’s disease (Kim et al. 2012).
Multiple Hereditary Exostoses (Multiple Osteochondromatosis)
Multiple hereditary exostoses are a rare autosomal dominant condition that has numerous cartilage-capped benign bony lesions, osteochondromas, at areas of active bone growth. The incidence is approximately 1 in 50,000 (Vanhoenacker et al. 2001). The osteochondromas typically affect both the upper and lower extremities (Fig. 6), with the humerus being the most common location in the upper extremity (Shapiro et al. 1979). Complications arising from this disorder include bony and cosmetic deformities; pathological fracture; bursa formation with subsequent pain; impingement of nearby tendons, nerves, and blood vessels; and malignant transformation (Murphey et al. 2000). Malignant transformation has been reported to occur in 3–5 % of patients with multiple hereditary exostoses (Murphey et al. 2000). It is impossible to remove all of the lesions, and therefore, only the symptomatic ones are addressed. Surgical excision is reserved for the lesions that cause growth disruption, pain, or neurovascular injury. Patients may require multiple procedures over the course of their lives to correct limb-length discrepancies or to excise painful lesions (McBride 1988).
Fig. 6
MHE involving bilateral upper and lower extremities (Courtesy of Shriners Hospital for Children)
Non-ossifying Fibroma
Non-ossifying fibroma is a benign fibrous growth resulting from a developmental defect in which fibrous connective tissue fills areas that normally ossify. These lesions develop in childhood and adolescence and generally arise in the metaphysis of long bones (Noh et al. 2013). The incidence of non-ossifying fibroma in skeletally immature children is 30–40 %, with males affected twice as much as females (Hudson et al. 1993; Shimal et al. 2010).
Pathoanatomy and Applied Anatomy Relating to Non-ossifying Fibroma
Non-ossifying fibromas are most commonly seen in the metaphysis of long bones. These lesions initially arise from the metaphysis and migrate toward the diaphysis with skeletal growth. Therefore, the lesions can be juxtacortical or even present within the cortex as the child ages.
Assessment of Non-ossifying Fibroma
Signs and Symptoms of Non-ossifying Fibroma
Non-ossifying fibromas are typically asymptomatic and discovered only incidentally. However, with minor trauma, pathological fractures can occur, causing pain.
Non-ossifying Fibroma Imaging and Other Diagnostic Studies
Radiographs of non-ossifying fibromas demonstrate lucent, cortical lesions with a well-circumscribed sclerotic rim. The lesion is typically eccentrically located and may be uni- or multiloculated (Schwartz and Ramos 1980; Howlett et al. 1998). Diagnosis of non-ossifying fibroma can be made on plain radiographs with an accuracy of 100 % (Schwartz and Ramos 1980).
Injuries Associated with Non-ossifying Fibroma
Non-ossifying fibromas rarely cause health problems due to the fact that they commonly disappear and are replaced with normal bone. If a large enough lesion weakens the bone, however, pathological fractures can occur (Noh et al. 2013).
Non-ossifying Fibroma Treatment Options
Nonoperative Management of Non-ossifying Fibroma
Indications/Contraindications
Observation is the mainstay of treatment for non-ossifying fibromas, as they are generally asymptomatic and commonly regress spontaneously. Contraindications to nonoperative treatment include symptomatic lesions, lesions larger than 3 cm and involving greater than 50 % of the bone’s diameter, or atypical-appearing lesions (Betsy et al. 2004).
Techniques
Small, asymptomatic lesions discovered incidentally do not require further follow-up unless the child experiences pain in that area.
Outcomes
Non-ossifying fibromas typically resolve spontaneously during adolescence and remain asymptomatic (Biermann 2002).
Operative Treatment of Non-ossifying Fibroma
Indications/Contraindications
Indications for surgical treatment of non-ossifying fibromas include symptomatic lesions causing pain or functional impairment, lesions larger than 3 cm and involving greater than 50 % of the bone’s diameter, and atypical-appearing lesions. Curettage and bone grafting is the mainstay of operative management (Noh et al. 2013).
Surgical Procedure
Preoperative Planning
The pathologist needs to be aware that the procedure is being performed and that a frozen section is being sent. Additionally, graft material needs to be readily available.
Positioning
Patients are placed supine on the operating room table and a radiolucent hand table is utilized.
Surgical Approach(es)
A direct approach to the tumor is utilized following the principles of tumor surgery including the utilization of a longitudinal incision.
Technique
Curettage and grafting is performed under fluoroscopic guidance utilizing curettes. It is essential to ensure that the physis is not damaged.
Preferred Treatment
Via a 2–3 cm incision, blunt dissection is carried down to bone. A small corticotomy is made to excise a window of bone while making a square with a drill bit and then connecting the “dots” with an osteotome. This is performed under fluoroscopic guidance to ensure the location is correct. Curettes are then utilized to curettage the lesion cavity. Following a thorough curettage, the defect is grafted and then the window of bone previously removed in replaced.
Surgical Pitfalls and Prevention
It is important to ensure that the entire lesion is curettaged to increase the chances of the lesion healing.
Management of Complications
Persistence of the cyst following surgical intervention requires a second operation to curettage and graft the lesion.
Osteoblastoma
Osteoblastoma is a rare benign bone tumor accounting for approximately 1 % of bone tumors (Cerase and Priolo 1998). These lesions commonly present in the second decade of life and affect males more often than females.
Pathoanatomy and Applied Anatomy Relating to Osteoblastoma
Osteoblastomas are generally larger than 2 cm in diameter and are most often located in the axial skeleton (Cerase and Priolo 1998). The tumor is comprised of osteoid and woven bone, and therefore, its histology is virtually identical to that of an osteoid osteoma. Fortunately, the location and size of the lesion can aid in differentiating between an osteoblastoma and an osteoid osteoma. Osteoblastomas are typically located in the medullary portion of bones, whereas osteoid osteomas can be located in the cortical, medullary, or subperiosteal region of bones.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


