Chapter 525 Bartter and Gitelman Syndromes and Other Inherited Tubular Transport Abnormalities
525.1 Bartter Syndrome
Rajasree Sreedharan and Ellis D. Avner
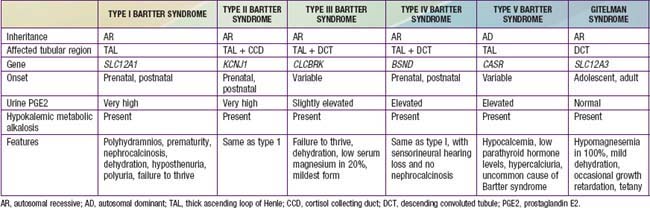
Bartter syndrome is a group of disorders characterized by hypokalemic metabolic alkalosis with hypercalciuria and salt wasting (Chapter 52) (Table 525-1). Antenatal Bartter syndrome (types I, II, IV) (also called hyperprostaglandin E syndrome) typically manifests in infancy and has a more-severe phenotype than classic Bartter syndrome (type III), including maternal polyhydramnios, neonatal salt wasting, and severe episodes of recurrent dehydration. The milder phenotype, classic Bartter syndrome, manifests in childhood with failure to thrive and a history of recurrent episodes of dehydration. A phenotypically related disease, Gitelman syndrome has a distinct genetic defect and is discussed in Chapter 525.2 (see Table 525-1). One distinct variant of antenatal Bartter syndrome is associated with sensorineural deafness (type IV).
Pathogenesis
The biochemical features of Bartter syndrome, including hypokalemic metabolic alkalosis with hypercalciuria, resemble those seen with chronic use of loop diuretics and reflect a defect in sodium, chloride, and potassium transport in the ascending loop of Henle. The loss of sodium and chloride, with resultant volume contraction, stimulates the renin-angiotensin II-aldosterone (RAA) axis. Aldosterone promotes sodium uptake and potassium secretion, exacerbating the hypokalemia. It also stimulates hydrogen ion secretion distally, worsening the metabolic alkalosis. Hypokalemia stimulates prostaglandin synthesis, which further activates the RAA axis. Bartter syndrome has been associated with 5 distinct genetic defects in loop of Henle transporters (see Table 525-1). Each contributes, in some manner, to sodium and chloride transport. Mutations in the genes that encode the Na+/K+/2Cl− transporter (NKCC2, the site of action of furosemide), the luminal potassium channel (ROMK), combined chloride channel (CLC-Ka, CLC-Kb), or subunit of chloride channels (barttin) cause neonatal Bartter syndrome. Isolated defects in the genes that produce the basolateral chloride channel ClC-Kb cause classic Bartter syndrome.



