Group
Type of involvement
Group 1
Mainly limb involvement (four limbs, lower limbs or upper limbs)
Group 2
Limbs are affected with involvement of other parts of the body
Group 3
Limbs are affected with central nervous system involvement
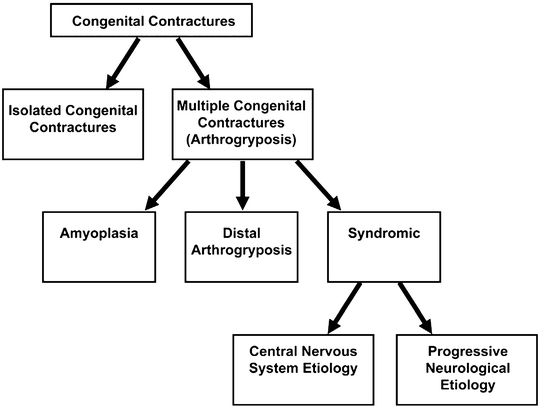
Fig. 18.1
Types of congenital contractures [4]. Reproduced with permission from Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: a review and update. J Bone Joint Surg Am. 2009 Jul;91 Suppl 4:40–6
Group 1: Disorders Affecting Mainly the Four Limbs
The most common disorder of this group is amyoplasia (a = no, myo = muscle, plasia = growth) meaning no muscle growth. It is the most common form of arthrogryposis, and represents one third of all cases. Most affected individuals have all four limbs involved. It has not been observed to recur in siblings [1].
Distal arthrogryposes (DA), as the name indicates, involve the distal joints, and do not involve a primary neurological and/or muscle disease. Distal arthrogryposes share a pattern of hand and foot involvement, limited involvement of proximal joints, and variable expressivity [4]. These disorders are inherited as autosomal dominant traits. Currently, distal arthrogryposes are subdivided into 10 types, depending on the number and nature of additional features [4]. Of these, type 5 can be further divided into several subtypes based on additional phenotypic features [8, 9]. Type I (DA1) is characterized by a typical positioning of the hands with medially overlapping fingers, clenched fists, ulnar deviation of the fingers and camptodactyly, and clubfoot. The degree of joint deformity is highly variable. The other types of distal arthrogryposes involve other body parts (e.g., eyes, mouth), and hence are presented as part of group 2 arthrogryposes.
There are other types of arthrogryposis involving mainly the limbs and which are included in group 1, such as contractural arachnodactyly and symphalangism [1].
Group 2: Disorders Affecting the Limbs with Involvement of Other Parts of the Body
Distal arthrogryposes (DA) other than type 1 are included in this group as in addition to contractures of the hands and feet, they involve other body parts. Type 2 can be further separated into two subtypes, Freeman-Sheldon syndrome (FSS or DA2A) and Sheldon-Hall syndrome (SHS or DA2B) [4]. The defining characteristic in FSS is that oropharyngeal abnormalities, scoliosis, and a very small oral orifice, puckered lips, and dimple in the chin, hence the name “whistling-face syndrome” (Fig. 18.2). SHS has similar congenital contractures present in DA1, with the addition of more prominent nasolabial folds, palpebral fissures, and a small mouth [4]. DA3 and DA4 are very rare. DA5 is unique in that ocular abnormalities are present in addition to muscle contractures. Other types of DA are very rare and have distinguishing features, such as very short stature and cleft palate in DA6, and inability to fully open the mouth and pseudocamptodactyly in DA7 [4].

Fig. 18.2
Photograph of a toddler with Freeman-Sheldon syndrome showing typical facies. Reproduced with permission from Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: a review and update. J Bone Joint Surg Am. 2009 Jul;91 Suppl 4:40–6
In addition to DA, there are many different syndromes involving muscle contractures and other body areas [2], such as multiple pterygium syndromes, diastrophic dysplasia, and Larsen syndrome. Multiple pterygium syndromes involve a web or triangular membrane that forms across a joint [2]. Different pterygium syndromes have different forms of inheritance and features. Popliteal pterygium syndrome is the most common of the multiple pterygium syndromes, but is very rare and affects 1 in 300,000 live births. It is inherited as autosomal dominant with a wide pattern of expression. Key features include genitourinary, craniofacial, and extremity malformations in association with popliteal webs that vary greatly in severity. The fixed knee and ankle deformities render ambulation difficult (Fig. 18.3). Diastrophic dysplasia involves contractures of the shoulders, elbows, interphalangeal joints, and hips [1, 2]. This type of dwarfism has an autosomal recessive inheritance. Other features include cystic masses involving the external ear and cleft palate [1]. Mortality rate is increased during infancy, but life expectancy is normal past infancy. Larsen syndrome involves a distinctive facial appearance with flattening of the face, prominent forehead, depressed nasal bridge, widely spaced eyes, and ligamentous hyperlaxity, as well as flexed hips and hyperextended knees [1]. Often, there are multiple joint dislocations (hips, knees, shoulder, and elbows), hand deformities with long cylindrical fingers, feet deformities in the form of equinovarus or equinovalgus, and a high incidence of spine anomalies, specifically cervical kyphosis caused by marked hypoplasia of one or two vertebral bodies. Congenital cardiac and respiratory anomalies may also be present. The management of the orthopedic problems is usually surgical.
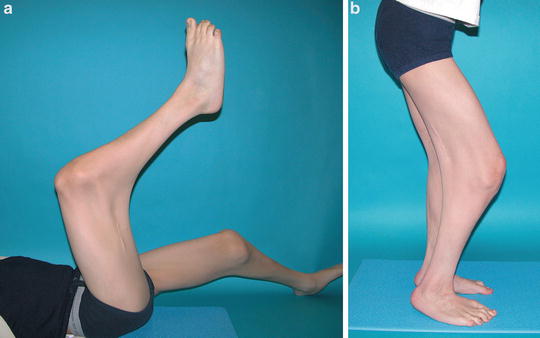
Fig. 18.3
(a, b) Eleven-year-old boy with popliteal pterygium syndrome
Group 3: Disorders Affecting the Limbs with Involvement of the Central Nervous System
Numerous syndromes with involvement of the central nervous system can have associated congenital contractures [2]. These may be caused by developmental abnormalities of the forebrain in utero, chromosomal deletions or rearrangements, or genetic mutations. These disorders are the most common cause of severe arthrogryposis [4]. Nerve conduction studies and EMG may be indicated in the case of neuromuscular disorders associated with arthrogryposis [4].
Genetic Aspects of Arthrogryposis
Multiple congenital contractures are present in a number of genetic syndromes. Many specific types of arthrogryposis have been mapped to loci in human chromosomes [1, 2]. The disorder may be caused by a single gene, in which case the disorder can be autosomal dominant, autosomal recessive, or X-linked. Rarely, disorders have a much higher risk of recurrence, such as mitochondrial inheritance. The presence of chromosomal abnormalities is prominent in cases of multiple congenital contractures with intellectual disability. Recent studies have identified several genetic mutations responsible for different forms of arthrogryposis [12–15]. Some types of arthrogryposis are sporadic, such as amyoplasia, in which genetics does not play a role in its inheritance. Prenatal testing and genetic counseling are indicated for parents with a higher risk of having another affected child.
Intellectual Skills
Intelligence is normal in many forms of arthrogryposis, including amyoplasia, distal arthrogryposis type I, diastrophic dysplasia, and Larsen syndrome. Patients with certain forms of arthrogryposis associated with chromosomal abnormalities or with CNS involvement may have decreased cognitive function [1].
Workup of a Child with Arthrogryposis
Any infant or child seen with multiple contractures should have a thorough clinical evaluation including a comprehensive family history, physical examination, and if necessary, further workup. In the physical examination, it should be determined first if arthrogryposis involves only the limbs or if it involves also other parts of the body, specifically the central nervous system. Second, it is important to determine which part of the limbs is involved, and to what extent. The range of motion of each joint should be carefully recorded. Radiographs of the spine, hips, and feet should be obtained. Bone mineral density appears to be lower in children with arthrogryposis compared to age-matched peers [16], and should be investigated.
It is essential to determine if this condition is genetic or nongenetic, not only for management purposes but also for counseling the parents on the risk of future pregnancies. Genetic investigations should include both blood cells and skin fibroblasts as, in some cases, mosaicism may exist (when chromosomal studies on blood cells are normal but an abnormality is detected in the skin fibroblasts). The most common form of arthrogryposis, amyoplasia, is not a hereditary condition and no specific genetic diagnosis can be made in this condition.
The focus of this chapter is on lower limb deformities as it pertains to amyoplasia, as it is the most common form of arthrogryposis.
Clinical Picture
A child with amyoplasia presents with a typical clinical picture. In the upper limbs, the shoulders are internally rotated and adducted; the elbows are extended, the forearms are pronated, the wrist and fingers are flexed (Fig. 18.4). This pattern of upper extremity involvement is often described as a policeman’s hand or a waiter’s tip hand (Fig. 18.5). In the lower limbs, the foot is most commonly affected (about 90 % of cases), followed by knee deformities (about 70 % of cases) and then hip deformities in about 40 % of patients. Flexion, abduction, and external rotation contractures of the hips are typically observed (Fig. 18.6) (Box 18.1). Often, there is loss of skin creases across the joints and dimpling at the sites of the joints. The muscles are severely atrophied. Children with amyoplasia have normal IQ and normal sensation [17].
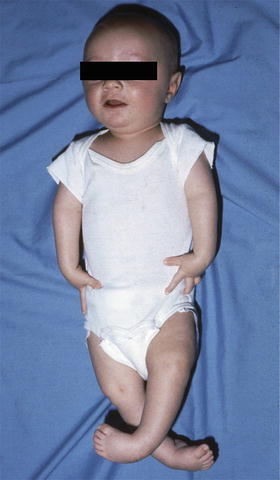
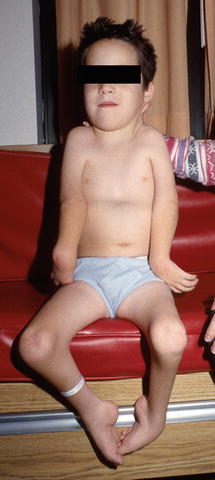

Fig. 18.4
Photograph of a 5-month-old baby with arthrogryposis showing bilateral clubfeet, extension contractures of the knees, and upper limb involvement with extended elbows and flexed wrists
Fig. 18.5
Photograph of a 5-year-old child with arthrogryposis showing typical upper and lower limb deformities, including policeman tip position with flexed wrist and extended elbows in the upper limb, clubfeet and knee flexion contractures in the lower limb
Fig. 18.6
Photograph of a 2-year-old child showing typical flexion abduction and external rotation contractures of the hips
Box 18.1. Diagnostic Approach for the Lower Limbs in a Child with Amyoplasia
The deformities are present at birth.
All four limbs are usually affected.
Sensation is normal.
Muscular atrophy and weakness is present.
Normal intelligence.
The feet are most commonly affected.
Knee flexion contractures are more common than extension contractures.
Hip flexion, external rotation, and abduction contractures of the hip, hip subluxation, or dislocation may be present.
Prognosis
The functional long-term prognosis is usually very good for most patients with amyoplasia, unlike other neuromuscular conditions. The contractures are usually most severe at birth and then gradually improve with life. The overall good prognosis should be clearly explained to the parents. Long-term ambulatory status and functional outcome at skeletal maturity are not necessarily correlated with the severity of arthrogryposis at birth, warranting early intensive treatment and rehabilitation in these children [18, 19].
General Management
Early intervention, in the form of aggressive physiotherapy including stretching, joint mobilization, and range of motion exercises, as soon as possible after birth, can facilitate optimal long-term functional outcome [4, 17–19]. Surgical interventions may also be indicated during the first year of life, as outlined below. Regular and frequent follow-up (every few weeks) is recommended during the first year of life to monitor progression and plan surgical interventions. This is followed by regular visits, typically every 6 months. Children with arthrogryposis should be followed in a multidisciplinary clinic. The overall goal in the management of these children is to improve function and ambulation while fostering optimal development. It is often recommended that the foot deformities be treated first, followed by the knee and finally the hip deformities [17].
Lower Limb
Involvement of the lower limb is common in arthrogryposis [11], and presents a challenging problem to the pediatric orthopedist. Lack of joint motion may be due to inadequate muscle development, lack of normal and mobile skin and tissue, and contractures with fibrous connective tissue. These issues must be considered when devising the treatment plan, and include the affected joints (e.g., foot, knee, and hip) (Box 18.2). Range of motion of the knee and hip may be improved with treatment, thereby facilitating ambulation [20].
Foot
Clubfoot deformity or talipes equinovarus is the most common foot deformity in arthrogryposis and is present in over 90 % of infants with amyoplasia [21]. Talipes equinovarus is much more common and is more rigid than the standard idiopathic clubfoot. It may be associated with knee contractures and hip contractures, subluxation, or dislocation (Fig. 18.7).
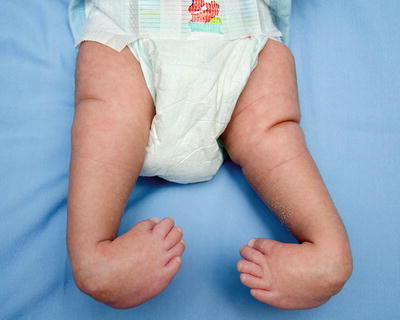
Fig. 18.7
Photograph of a 3-week-old infant with arthrogryposis showing right knee extension contracture, left knee subluxation, and bilateral clubfeet
The goal in the management of feet deformities is to obtain a painfree, plantigrade foot. Treatment should start immediately after birth with the Ponseti technique. Previous studies have reported that clubfeet in children with arthrogryposis are usually stiff and resistant to standard treatment and may require talectomy as a first-line treatment [22, 23]. However, more recently, it has been shown that tenotomy of the tendo Achilles before the start of the Ponseti technique may facilitate the manipulations and may yield good results with the Ponseti technique [24]. We recommend this approach as the initial treatment. In cases of failure of the Ponseti technique, a formal posteromedial release is recommended [25] even though the recurrence rate is high (Fig. 18.8). In cases where a posteromedial release is not sufficient to obtain a plantigrade foot, then a talectomy is advisable [22, 23]. In cases of recurrent deformities, repeat soft tissue releases could be dangerous as the anatomy is completely distorted and neurovascular damage may ensue. Consideration should be given to correction through multiple osteotomies. The use of gradual correction with external fixators is another viable option (Fig. 18.9). A standard Ilizarov circular fixator could be used [26] or a Taylor Spatial Frame with the foot program [27]. In patients near or at skeletal maturity presenting with residual or recurrent foot deformities, triple arthrodesis may be the best option [28].
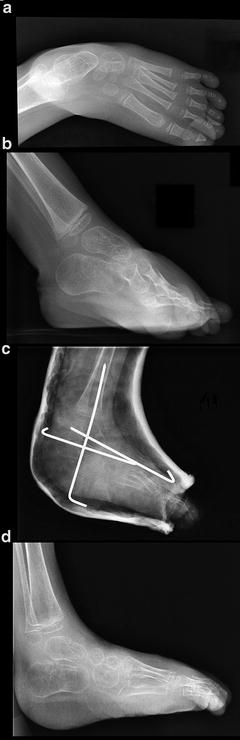
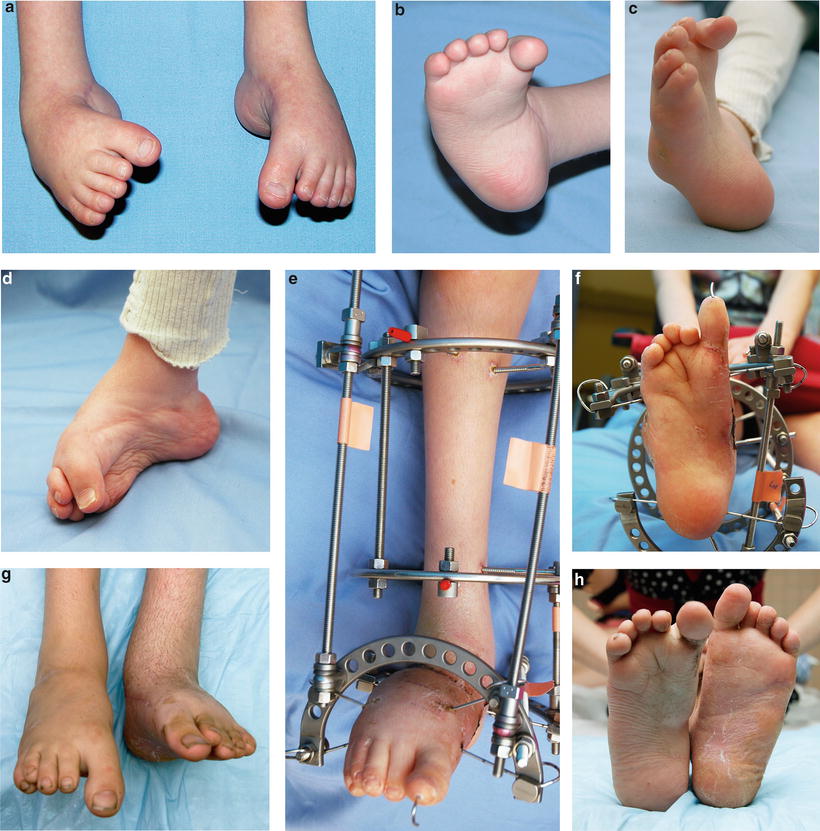
Fig. 18.8
(a, b) Radiographs of a 4-year-old child with arthrogryposis showing left clubfoot. (c) Postoperative radiograph following radical posteromedial release of the left foot after a failed Ponseti technique. (d) Three-month postoperative radiograph showing adequate correction
Fig. 18.9
Photographs (a, b) showing severe deformities of the right foot in a 3-year-old girl. The right foot is severely supinated at age 9 (c, d). Treatment of deformity with Ilizarov external frame (e, f). (g, h) Satisfactory alignment of the right foot post-Ilizarov removal at age 11
Conservative treatment in the form of the Ponseti technique, as described above, should be considered first. In case of recurrence or substantial residual deformity, a radical posteromedial release may be necessary around the age of 1 year followed by prolonged splinting to prevent or minimize recurrence of deformities [24, 27]. It should be clearly explained to the family that while such surgical treatment will eventually correct the deformity, the foot remains stiff, with a high likelihood of needing subsequent surgeries as the recurrence rate is very high [22, 29].
Congenital vertical talus is reported to occur in approximately 2–12 % of patients with arthrogryposis [30]. Because many of these patients have the potential to ambulate, the congenital vertical talus should be treated early, before walking age. Immediately after birth, the modified Ponseti technique should be initiated as described by Alaee, Boehm, and Dobbs [31]. This may be successful in the correction of the deformity. However, if the congenital vertical talus is rigid, then more aggressive treatment in the form of a one-stage release is usually recommended. A tibialis anterior transfer to the talar neck with or without Grice subtalar fusion is also recommended [32]. In very stiff feet and recurrent deformities, surgical options include excision of the navicular, talectomy, multiple osteotomies, and gradual correction with an external fixator (Fig. 18.10). If the patient is near skeletal maturity and has severe deformities, then a triple arthrodesis may be considered [32].
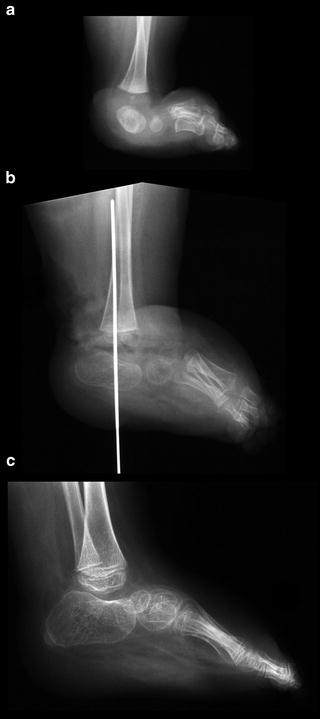
Fig. 18.10
(a) Radiograph showing vertical talus in a 2-year-old child with arthrogryposis. (b) Radiograph obtained after talectomy was performed shows good position of the calcaneus under the tibia. (c) Radiograph obtained 4 years postoperatively
Knee
The knee is the second most commonly affected lower extremity joint in amyoplasia (38–90 % of patients) [33]. Knee contractures can be the most disabling deformity as they prevent motion and anatomical limb alignment [34]. Among knee deformities, flexion contractures are the most common in arthrogryposis [35, 36] and are also more disabling than extension contractures [35]. The goal in the management of flexion deformities is to facilitate ambulation, sitting, and standing (Table 18.2). Knee deformities may be more difficult to treat than other deformities and, more importantly, present some of the most challenging problems causing gait disturbances and may render ambulation very difficult [33, 34]. Contrary to extension deformities that may not interfere with walking, flexion contractures of more than 20–30° usually interfere with ambulation and should be treated. In fact, the presence of knee flexion contractures is the single most important indicator of the potential for ambulation. We and others have shown that correction of knee flexion deformities has a direct positive impact on the ambulation potential of these children [19, 36]. In our own series of patients where knee flexion contractures were treated with supracondylar osteotomies (Fig. 18.11) or with gradual correction using an Ilizarov circular frame, we reported that ambulation gains were maintained in most patients despite some loss in correction with time in many patients [19]. However, quadriceps strength should also be taken into account in the management of knee deformities as quadriceps weakness (noted in about 60 % of patients) [37] may preclude any gains in ambulation despite correction of knee flexion deformity [33].
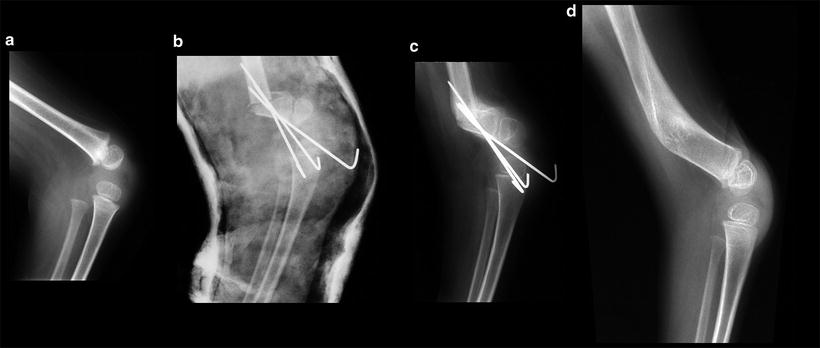
Table 18.2
Indications of management for knee contractures
Knee contracture severity | Degrees | Management |
|---|---|---|
Mild | 0–20 | • Physiotherapy • Stretching • Serial casting |
Moderate | 20–40 | • Anterior distal femoral hemiepiphysiodesis • Soft tissue release |
Severe | 40–60 | • Femoral extension osteotomy |
Very severe | 60+ | • Gradual distraction |
Fig. 18.11
(a) Preoperative radiograph showing a severe knee flexion deformity in a 5-year-old child with arthrogryposis. (b) Postoperative radiograph after supracondylar osteotomy was performed. (c) Radiograph obtained 2 months postoperatively. (d) Radiograph obtained 1 year postoperatively showing remodeling
The fact that there are several options for the treatment of knee flexion deformities emphasizes the challenge that these deformities present [34]. Treatment of knee deformities should start at birth with intensive stretching exercises and bracing. If the flexion deformity exceeds 30° and is not responsive to physiotherapy, serial casting or braces with hinges may be used in order to gradually stretch the contracture. If these conservative measures fail to decrease the contracture to 20° or less, more aggressive techniques should be used such as soft tissue releases, femoral supracondylar osteotomy with or without shortening, anterior hemiepiphysiodesis of the distal femur, and gradual correction of the deformity using external fixators. However, some of these techniques have a high rate of recurrence in the skeletally immature patient and the natural history of knee flexion deformity is typically one of progression [38].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


