Antiretroviral Pharmacology in Pregnant Women and Newborns
Mark Mirochnick
Brookie M. Best
Introduction
As infection with human immunodeficiency virus (HIV) continues to spread throughout the world, HIV/AIDS has become a major contributor to global morbidity and mortality. At the end of 2007, 33 million people worldwide were estimated to be living with HIV infection, of whom nearly half are women of childbearing age and 2.0 million are children under the age of 15 years, most of whom acquired HIV infection from mother-to-child transmission (1). Worldwide deaths from AIDS in 2007 were estimated at 2.0 million people, including 290,000 children. Although the number of new HIV infections worldwide likely peaked in the late 1990s at an estimated rate of 3.0 million people per year, in 2007 an estimated 2.7 million people were still newly infected, including 370,000 children (1). Those children who escape mother-to-child HIV transmission may still suffer from the AIDS epidemic. The number of children in sub-Saharan Africa who had lost one or both parents because of AIDS was estimated at 11.4 million in 2007 (1).
Indications for the use of Antiretrovirals in HIV-Infected Pregnant and Postpartum Women and their Neonates
Much progress has been made in recent years in treating HIV infection. Currently, more than 20 antiretroviral drugs in five classes are available in the United States (Table 22.1) and new drugs are in development. The use of combination regimens of three or more antiretrovirals, often referred to as highly active antiretroviral therapy (HAART), has resulted in dramatic improvements in HIV morbidity and mortality. However, while antiretroviral therapy improves the health and prolongs the lives of HIV-infected patients, it does not eradicate the virus or cure the infection. Once initiated, antiretroviral therapy generally continues for life. Because of the long asymptomatic phase that generally follows HIV infection, the lifelong duration of antiretroviral treatment, the side effects and toxicities of the drugs, and the frequent development of viral resistance resulting in loss of antiretroviral efficacy, clinical criteria have been established to determine when antiretroviral therapy should be initiated. Guidelines have been published outlining these criteria for the initiation of antiretroviral therapy in HIV-infected adults and children living in areas where the availability of medical resources permits wide-scale access to antiretroviral therapy and in resource poor areas with limited access (2,3,4). Both sets of adult guidelines recommend initiation of antiretroviral therapy in HIV-infected pregnant women according to the same criteria as those used in nonpregnant adults.
Antiretroviral therapy may also be used in pregnant women for prevention of mother-to-child HIV transmission in those women whose HIV disease is not sufficiently advanced to meet the clinical criteria for initiation of antiretroviral therapy. HIV can be passed from mother to child in three ways: across the placenta during pregnancy (transplacental), from exposure during labor and delivery to maternal blood and other bodily fluids (intrapartum), and from breast milk during nursing (5). In the absence of treatment, 15% to 45% of HIV-infected pregnant women will pass the infection on to their infants, with 5% to 10% of infants born to HIV-infected women infected across the placenta, 10% to 20% infected from exposure at or around the time of delivery, and 10% to 20% infected from breast milk (5).
Effective therapeutic strategies to prevent mother-to-child HIV transmission have been developed. In 1994, the PACTG 076 protocol demonstrated that administration of a zidovudine regimen composed of oral dosing initiated at 14 to 34 weeks’ gestation, continuous intravenous infusion during labor, and 6 weeks of oral dosing to the newborn reduced mother-to-child transmission by 67% (transmission rate of 7.6% with zidovudine compared with 22.6% with placebo) (6). The relative importance of the three components (prenatal, intrapartum, and postpartum) of the PACTG 076 regimen in reducing mother-to-child transmission has not been fully defined. A retrospective analysis of the use of abbreviated zidovudine regimens in New York State found that zidovudine was most effective if
initiated during pregnancy, reducing transmission from 26.6% to 6.1%, and had decreased efficacy if begun during labor or within 48 hours after birth, reducing transmission to 9% to 10% (7).
initiated during pregnancy, reducing transmission from 26.6% to 6.1%, and had decreased efficacy if begun during labor or within 48 hours after birth, reducing transmission to 9% to 10% (7).
Table 22.1 Pregnancy Dosing Recommendations for anti-HIV Drugs Available for Use in the United States | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Further reductions in mother-to-child HIV transmission have been associated with the use of elective cesarean delivery performed before rupture of membranes and of combination antiretroviral regimens. Elective cesarean reduces the rate of mother-to-child transmission to 2% or less when used in combination with zidovudine (8,9). Administration of combination regimens of multiple antiretrovirals during pregnancy is associated with reductions in the rate of mother-to-child transmission to 1.5% or less in women who do not breast-feed their infants (10,11,12). Guidelines for the use of antiretroviral drugs to prevent mother-to-child transmission have been developed (13). Although some variation exists among practitioners, the use of combination antiretroviral regimens during pregnancy, elective cesarean section if the HIV RNA level at the time of delivery is not suppressed to below the level of
assay detection, and formula feeding in place of breastfeeding have become standard treatment of HIV-infected pregnant women in areas of the world where medical resources are sufficient to make these interventions readily available.
assay detection, and formula feeding in place of breastfeeding have become standard treatment of HIV-infected pregnant women in areas of the world where medical resources are sufficient to make these interventions readily available.
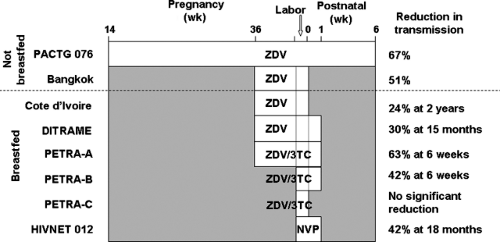 Figure 22.1. Duration of antiretroviral therapy and reduction in mother-to-child HIV transmission. White bar indicates duration of prenatal, intrapartum and/or postnatal dosing. ZDV, zidovudine; 3TC, lamivudine; NVP, nevirapine. From PACTG 076, Wade et al. (7); Bangkok, Shaffer et al. (14); Cote d’Ivoire, Wiktor et al. (15); DITRAME, Dabis et al. (16); PETRA, PETRA Study Team (17); HIVNET 012, Jackson et al. (19). |
Unfortunately, those parts of the world most affected by HIV generally lack sufficient health care resources to make these interventions widely available. Less intensive antiretroviral regimens more practical for use in resource poor areas have been developed (3,5) (Fig. 22.1). Shortened zidovudine regimens that start at 36 to 38 weeks of pregnancy, use oral rather than intravenous dosing during labor, and decrease or eliminate postnatal infant dosing have been shown to reduce transmission by 38% to 50%, compared with the 67% reduction seen with the full PACTG 076 regimen (14,15,16). A short-course regimen combining zidovudine and lamivudine (arm A of the PETRA study—maternal treatment from 36 weeks’ gestation through delivery, infant treatment for 1 week after birth) reduced transmission at 6 weeks after birth by 63% compared with placebo (17). Arm B of that study (zidovudine and lamivudine to the mother during labor and to the infant for 1 week after birth) reduced transmission at 6 weeks by 42%, while arm C (intrapartum dosing only) had no impact on transmission (17).
The least intensive, lowest cost antiretroviral regimen shown to be effective in preventing mother-to-child HIV transmission involves two doses of nevirapine—one oral dose to the mother during labor and another oral dose to the infant at 48 to 72 hours after birth. This nevirapine two-dose intrapartum–postnatal regimen reduced mother-to-child HIV transmission by 41% in a breastfeeding population (18,19). In a South African study, mother-to-child transmission at 8 weeks of age was equally low (9% to 12%) with either an abbreviated zidovudine–lamivudine regimen based on arm B of the PETRA study or intrapartum–postnatal nevirapine (20).
In a Thai study, the combination of third trimester zidovudine with intrapartum–postnatal nevirapine and formula feeding resulted in a mother-to-child HIV transmission rate below 2%, nearly equivalent to that seen with the use of HAART during pregnancy (21). The combination of zidovudine starting at 28 weeks’ gestation plus intrapartum–postnatal nevirapine is currently recommended by WHO as the first choice antiretroviral regimen for use in resource-constrained settings (22).
In resource-constrained areas where formula feeding is not safe or affordable, prevention of mother-to-child HIV transmission via breast milk remains a major problem. Breastfeeding is the cultural norm in most resource-constrained settings and is critical to infant survival. In a meta-analysis, breastfeeding was associated with a sixfold decrease in mortality due to infectious diseases for infants younger than 2 months (23). If a breastfeeding mother is HIV infected, her infant is at risk of HIV infection from breast milk, with an estimated risk of transmission of 0.6% to 0.9% for every month of breastfeeding (24,25). Breast milk HIV transmission is reduced but not eliminated with exclusive breastfeeding, as opposed to mixed feeding with formula, water, juice, and other liquids or solids in addition to breast milk (26). Preliminary studies of the use of antiretrovirals to prevent breast milk HIV transmission have been reported. Treatment of the lactating mother with combination antiretroviral therapy and the nursing infant with nevirapine both seem to protect against infection of the infant (27,28,29).
Effect of Pregnancy on Drug Disposition
Maternal physiologic changes associated with pregnancy may have a considerable impact on drug disposition. All four components of drug disposition—absorption, distribution, metabolism, and excretion—may be affected by
pregnancy. When antiretrovirals are used during pregnancy, whether for prevention of mother-to-child HIV transmission or treatment of the mother’s underlying HIV infection, or both, special consideration must be taken and usual adult dosing may need to be modified.
pregnancy. When antiretrovirals are used during pregnancy, whether for prevention of mother-to-child HIV transmission or treatment of the mother’s underlying HIV infection, or both, special consideration must be taken and usual adult dosing may need to be modified.
Pregnancy results in significant changes in gastrointestinal function that may impact drug absorption. Nausea and vomiting, especially pronounced in early pregnancy, may decrease drug absorption. Plasma progesterone increases during pregnancy, associated with a 30% to 50% decrease in intestinal motility and increases in gastric emptying and intestinal transit times (30). The ionization and absorption of weak acids and bases may be affected by increased gastric pH due to a 40% reduction in acid secretion (31). Although these physiologic changes would be expected to result in delayed drug absorption and reduced peak maternal blood concentrations, few studies have evaluated the clinical impact of these changes in a rigorous manner (32).
The profound changes in body composition during pregnancy are well known. During an average pregnancy, total body water increases by 8 L, plasma volume enlarges by 50%, and body fat stores increase, changing volume of distribution of both hydrophilic and lipophilic drugs (33). The dilutional decrease in serum albumin as well as competitive inhibition from steroid hormones results in decreased protein binding (34). As a result of these physiologic changes, volume of distribution generally increases and peak drug concentrations decrease during pregnancy. The decrease in protein binding also results in an increase in the free fraction, or unbound fraction, of drug. Unbound drug is the pharmacologically active drug moiety, available for binding to sites of action and for biotransformation and elimination. The free fraction of many drugs, including theophylline, diazepam, salicylates, and some β-lactam antibiotics, have been shown to increase during pregnancy as a result of changes in protein binding (35,36,37).
The effect of pregnancy on drug elimination is variable but may be significant. Progesterone may induce hepatic drug metabolic pathways as has been shown for phenytoin (38). Drug metabolism may be reduced due to competition with estrogen and progesterone for metabolic binding sites as has been demonstrated for theophylline and caffeine (39). Renal function increases during pregnancy, with 25% to 50% increases in renal plasma flow and GFR. Clearance of renally excreted drugs, such as ampicillin and gentamicin, and drug metabolites increases during pregnancy (40,41).
Although the disposition of most drugs will be measurably changed by the physiologic changes of pregnancy, the need for dosing adjustment will be determined by the magnitude of these changes and the pharmacokinetic–pharmacodynamic relationship for each drug. Multiple and contradictory effects may coexist (31). Absorption often decreases and volume of distribution and clearance increase, leading to decreased total plasma concentrations, while protein binding decreases, leading to a larger free fraction of drug. Although rigorous pharmacokinetic studies are rare in pregnant women, clinical experience suggests that these effects may be significant. Between one quarter and one-third of pregnant women with epilepsy will have an increase in seizure frequency during pregnancy associated with subtherapeutic drug levels (42). Pregnant women receiving antidepressants often develop increased depressive symptoms and require dose increases to maintain adequate serum drug concentrations during late pregnancy (43). Pharmacokinetic studies are difficult to perform during pregnancy and, until recently, women of reproductive age were excluded from most clinical trials. As a result, published studies of drug disposition in pregnancy are limited, often contradictory and almost always fail to provide clinically relevant guidelines (44). Monitoring of drugs with narrow therapeutic indices, such as phenytoin and theophylline, has been recommended during pregnancy (45). However, routine laboratory drug assays must be interpreted with caution during pregnancy, as they measure total drug concentration, not the free fraction, and therapeutic and toxic effects may occur at lower total concentrations in the face of decreased protein binding.
Safety of Antiretrovirals During Pregnancy
The current approach to prevention of mother-to-child HIV transmission through the use of antiretrovirals can reduce the rate of mother-to-child HIV transmission below 2% (10,12). The vast majority of fetuses exposed to antiretrovirals during pregnancy will not be infected with HIV, making the safety of these agents for both fetus and mother of paramount importance. The risk or safety of drugs used in pregnancy is difficult to assess. Preclinical drug evaluations include in vitro and animal in vivo studies for carcinogenicity, mutagenicity, and reproductive and teratogenic effects. Direct extrapolation of the results of these preclinical studies to humans is of uncertain value. Of approximately 1,200 known animal teratogens, only about 30 have been shown to be teratogenic in humans (46). However, in at least one case, that of isotretinoin, animal studies demonstrating severe teratogenicity prevented widespread use of this agent in pregnant women and averted a likely epidemic of birth defects (47). Preclinical animal studies are especially concerning for efavirenz, zalcitabine, delavirdine, and tenofovir (see descriptions of the pharmacology of individual antiretroviral agents later) and if possible these agents should be avoided during pregnancy, especially during the first trimester.
Human perinatal phase I, II, and III antiretroviral studies are too small and of too limited duration to adequately assess for adverse effects, especially those effects that are uncommon or first appear outside of infancy. The largest amount of data is available for zidovudine, which has been consistently associated with transient neonatal anemia but otherwise appears to be without harmful effects following in utero and postnatal exposure (48,49). Safety data from perinatal clinical trials specific to individual antiretroviral drugs are included in the descriptions of the pharmacology of individual antiretroviral agents below.
The potentially life-threatening consequences of HIV infection to mother and fetus have compelled the use of many antiretroviral agents, often as part of combination regimens, in pregnant women and their newborns in the absence of definitive safety data. Monitoring of this clinical
experience is ongoing in epidemiological cohort studies and in the Antiretroviral Pregnancy Registry, a postmarketing surveillance registry sponsored by the pharmaceutical industry to collect information about major teratogenic effects with exposure to 18 antiretroviral agents (http://www.apregistry.com/). Although these cohorts will include large numbers of exposures and provide useful information, the data include exposure to a large variety of drug combinations, making it difficult to establish safety assessments for individual agents. These monitoring studies are also limited by ethnic, social, and clinical differences in the populations studied, the lack of randomized comparator groups, inadequate information about confounding variables, and uncertain accuracy of outcomes (50).
experience is ongoing in epidemiological cohort studies and in the Antiretroviral Pregnancy Registry, a postmarketing surveillance registry sponsored by the pharmaceutical industry to collect information about major teratogenic effects with exposure to 18 antiretroviral agents (http://www.apregistry.com/). Although these cohorts will include large numbers of exposures and provide useful information, the data include exposure to a large variety of drug combinations, making it difficult to establish safety assessments for individual agents. These monitoring studies are also limited by ethnic, social, and clinical differences in the populations studied, the lack of randomized comparator groups, inadequate information about confounding variables, and uncertain accuracy of outcomes (50).
Published reports of pregnancy outcomes associated with perinatal antiretroviral exposure highlight these difficulties. An initial observational study from Switzerland reported in 1998 on pregnancy outcome in 37 women receiving combination antiretroviral therapy, including 16 women receiving protease inhibitors. Twenty-nine women had adverse events, including preterm delivery in 10 (51). However, a meta-analysis published in 1998 that examined the association of maternal HIV infection and perinatal outcomes in 31 studies found that the risk of premature delivery was increased 1.83-fold among HIV-infected women (52). A collaborative European group reported on 2,414 uninfected children born to HIV-infected mothers between 1985 and 2001, of whom 1,008 were exposed to antiretroviral agents during pregnancy, delivery, and/or the neonatal period (53). No pattern or prevalence of congenital anomalies or low birth weight was associated with antiretroviral exposure, but exposure to any antiretroviral was associated with mild transient anemia in early life. In an updated 2004 report, this group reported that in 4,372 HIV-infected European women delivering between 1986 and 2004, the risk of delivery at less than 37 weeks was 1.9-fold increased with HAART started during pregnancy and 2.1-fold increased with HAART started pre-pregnancy (54). In contrast, several US studies have shown no association between antiretroviral use and preterm delivery or other adverse pregnancy outcomes (55,56). A recent meta-analysis of 14 studies that examined the association between antiretroviral therapy during pregnancy and premature delivery found no increased risk of premature delivery associated with antiretroviral use (57). Although no clear explanation is available for the inconsistency between the European and US experience, the potential for an increased risk of premature birth in HIV-infected pregnant women receiving combination antiretroviral therapy during pregnancy should be recognized and included in clinical discussions of the risks and benefits of antiretroviral therapy (58).
Nucleoside reverse transcriptase inhibitors (NRTIs) are known to inhibit mitochondrial function (59). Their long-term use may result in toxicity associated with depletion of mitochondrial DNA, although these toxicities generally resolve with cessation of use (60). Investigators in France reported that 8 of 1,754 uninfected infants with in utero and postnatal nucleoside reverse transcriptase exposure had biopsy proven persistent mitochondrial dysfunction (61). All eight infants were exposed to zidovudine, four as zidovudine monotherapy and four in combination with lamivudine. Five, of whom two died, presented with delayed onset of neurological symptoms while three were symptom free but had laboratory abnormalities (61). This report stimulated a worldwide search for other uninfected infants with perinatal antiretroviral exposure and evidence of mitochondrial toxicity. One US infant has been reported who was exposed to antiretrovirals for the last 4 weeks of pregnancy and had severe neurological symptoms present at birth and a muscle biopsy confirming mitochondrial dysfunction (62). Otherwise no cases consistent with mitochondrial dysfunction could be found among the uninfected antiretroviral exposed children in several research cohorts, including 3 US cohorts totaling 19,486 children, 1 African cohort of 1,798 children, 1 Thai cohort of 330 children, and a European cohort of 1,008 children (53,63,64,65,66). Although the difficulty in finding cases fitting the description of mitochondrial dysfunction in these other cohorts from around the world is encouraging, the limitations in diagnosing mitochondrial dysfunction without prospective neurologic and laboratory evaluations prevents a definitive conclusion about the relationship between perinatal nucleoside exposure and persistent mitochondrial toxicity from being drawn (67).
The health of the mother must not be forgotten when evaluating the risks and safety of antiretroviral use during pregnancy. The physiologic changes of pregnancy may lead to toxicities unique to pregnancy, such as premature delivery, or may make the mother more susceptible to toxicities described in nonpregnant adults. Abnormalities in carbohydrate metabolism are common side effects in nonpregnant adults receiving combination antiretroviral regimens, especially those including protease inhibitors, raising concerns about an increase in gestational carbohydrate intolerance in pregnant women receiving antiretrovirals (68). However, several studies have shown no significant association between type of antiretroviral treatment and gestational diabetes, including a recent prospective study that performed detailed evaluations for glucose intolerance and insulin resistance in HIV-infected pregnant women receiving protease-inhibitor–containing and nonprotease-inhibitor–containing regimens (69,70). Pregnancy may also predispose to mitochondrial dysfunction associated with nucleoside exposure. Lactic acidosis and hepatic steatosis are rare nucleoside toxicities attributed to mitochondrial dysfunction from nucleoside analogue exposure (71). Three fatal and several less severe cases of lactic acidosis and hepatic steatosis have been reported in pregnant or postpartum women whose antiretroviral regimen during pregnancy included stavudine and didanosine (72,73). Acute fatty liver and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets), two rare but life-threatening syndromes that occur during pregnancy, have been associated with abnormal mitochondrial fatty acid oxidation in mother and/or fetus (74). Pregnancy may possibly predispose to the mitochondrial dysfunction leading to all three syndromes—acute fatty liver of pregnancy, HELLP syndrome, and nucleoside-associated lactic acidosis/hepatic steatosis. The combination of stavudine and didanosine should be used with caution during pregnancy and only when other drug combinations have failed.
Another risk to the mother from antiretroviral use during pregnancy is the development of viral resistance. Drug-resistant HIV mutants develop when suppression of viral replication is incomplete during antiretroviral therapy, so that ongoing viral replication and genetic mutation leads to the selection of drug-resistant HIV variants (75). Complete viral suppression is achieved in only 40% to 80% of patients beginning HAART regimens and is rare in patients receiving single or dual drug therapy (76,77). Although drug-resistant HIV strains fade after treatment with an individual drug ceases, they generally recur with reexposure to the drug, limiting the efficacy of that antiretroviral agent in the future.
HIV drug resistance develops most easily for the first-generation non-NRTIs, requiring only a single mutation of the HIV genome (78). In individuals with uncontrolled viral replication, instability of the HIV genome results in daily production of single mutation nevirapine-resistant virus even in the absence of nevirapine therapy (79). Nevirapine has a long half-life and can be detected in plasma up to 3 weeks after administration of a single intrapartum dose (80). Under the selective pressure of nevirapine monotherapy, resistant viral strains proliferate and become detectable 1 to 2 weeks after initiation of treatment (79). Among women exposed to a single dose of nevirapine during labor for prevention of mother-to-child transmission, nevirapine-resistant HIV isolates could be detected 6 weeks after delivery in 20% of women receiving no other antiretrovirals and in 15% of women receiving standard perinatal antiretroviral regimens (81,82). The clinical implications of the presence of nevirapine-resistant HIV are unclear. Drug-resistant HIV mutants tend to be less fit than wild-type virus and gradually disappear as wild-type virus reemerges once the selective pressure from continued antiretroviral exposure is removed. Nevirapine-resistant virus could not be detected 1 to 2 years later in any of 11 women who received a single intrapartum nevirapine dose and had detectable nevirapine-resistant HIV shortly after delivery (83). In women who received a second or third course of single-dose intrapartum nevirapine in subsequent pregnancies, resistant virus was detected in 38% within 1 year after delivery but in only 7% at 3 years after exposure (84). Single-dose nevirapine appears to be as effective in preventing HIV transmission in subsequent pregnancies as when it is used for the first time (85,86). Several studies have suggested that efficacy does not decrease when nevirapine-based combination therapy is started at least 6 to 12 months after delivery (87,88,89,90). The development of nevirapine resistance can be reduced by administration of a short course of antiretrovirals to the mother after delivery (91,92).
Resistance to zidovudine and protease inhibitors requires multiple mutations and develops more slowly than resistance to the first-generation non-NRTIs (93). In PACTG 076, antenatal zidovudine treatment resulted in low-level genotypic resistance in only 1 of 39 women tested (94). Follow-up of women enrolled in PACTG 076 over 4 years could find no differences in the clinical course or in the incidence of zidovudine-resistance mutations in the women who received zidovudine when compared with those who received placebo (95). In contrast, the development of resistance to the NRTI lamivudine requires only a single mutation, and four of five women receiving dual therapy with lamivudine and zidovudine during pregnancy developed lamivudine resistance by the time of delivery (96). In a larger study, lamivudine resistance was detectable 6 weeks after delivery in 52 of 132 women receiving lamivudine and zidovudine during pregnancy (97).
Drug-resistant HIV may be transmitted from mother to infant. In a group of 91 HIV-infected infants born in New York State in 1998 to 1999, 12.1% were infected with drug-resistant HIV strains (98). However, the presence of resistance mutations does not increase the rate of transmission (58). All pregnant women starting antiretroviral therapy or those on therapy with detectable HIV viral RNA levels should have resistance testing performed to ensure selection of an optimal regimen to suppress HIV viremia and minimize the risk of transmission (2,13).
Pharmacology of Individual Antiretroviral Agents
Nucleoside/tide Reverse Transcriptase Inhibitors
The first antiretroviral agents developed were NRTIs. Structurally similar to endogenous deoxynucleosides, these agents are prodrugs that are inactive until metabolized within the cell to triphosphorylated forms, which then inhibit HIV reverse transcriptase and viral DNA replication (99). Quantification of the intracellular concentrations of these active triphosphorylated forms is difficult, requiring large blood samples and sophisticated assay techniques (100,101). The half-life of these active intracellular metabolites generally exceeds that of the parent drug in plasma (99). Extracellular plasma NRTI concentrations do not correlate well with concentrations of the active intracellular forms, and the plasma pharmacokinetics of NRTIs does not generally correlate with therapeutic effect (102). Tenofovir, the first nucleotide to be approved for use against HIV, is an acyclic analogue of adenosine monophosphate that requires only two phosphorylation steps to reach its active form, tenofovir diphosphate (103).
Prolonged treatment of HIV-infected individuals with either a single agent in this class (monotherapy) or two agents (dual therapy) generally results in only a transient suppression of HIV replication, as resistance mutations develop in the HIV gene that codes for reverse transcriptase. As a result, NRTIs are recommended to be used only as part of a combination regimen of at least three drugs, most commonly two NRTIs with either a protease inhibitor or a non-nucleoside reverse transcriptase inhibitor, when treating HIV infection (2). Less intensive regimens in pregnant women have been shown to protect against mother-to-child HIV transmission, as described earlier.
Zidovudine
Zidovudine, the first antiretroviral to be developed, was also the first drug shown to prevent mother-to-child transmission (6). Zidovudine remains a common component of initial combination regimens to treat HIV infection and of regimens to prevent mother-to-child transmission (4). Absorption of zidovudine is rapid and complete. In nonpregnant adults receiving zidovudine oral doses of
200 mg, the average maximum concentration (Cmax) is 1.0 μg per mL reached at an average time of maximum concentration (Tmax) of 0.65 hours (104). Zidovudine is avidly metabolized by the liver by glucuronidation and undergoes extensive first pass metabolism, so that bioavailability averages 63% despite nearly complete absorption (105). Zidovudine is rapidly eliminated from the body, primarily by renal excretion as glucuronide (106). Zidovudine half-life (t1/2) averages around 1.1 hours and oral clearance (Cl/F) around 1.3 L per hour per kg in nonpregnant adults (107).
200 mg, the average maximum concentration (Cmax) is 1.0 μg per mL reached at an average time of maximum concentration (Tmax) of 0.65 hours (104). Zidovudine is avidly metabolized by the liver by glucuronidation and undergoes extensive first pass metabolism, so that bioavailability averages 63% despite nearly complete absorption (105). Zidovudine is rapidly eliminated from the body, primarily by renal excretion as glucuronide (106). Zidovudine half-life (t1/2) averages around 1.1 hours and oral clearance (Cl/F) around 1.3 L per hour per kg in nonpregnant adults (107).
Several studies have evaluated zidovudine pharmacokinetics during pregnancy. In early studies, zidovudine Cmax, Tmax, bioavailability, and t1/2 appeared no different than in historical values from nonpregnant adults (108,109). In two later studies where pharmacokinetics was compared during pregnancy and at 1 to 4 weeks postpartum in the same women, average Cl/F significantly increased during pregnancy by 47% to 65% while average area under the plasma concentration-time curve (AUC) decreased by 34% to 39% (110,111). The clinical significance of the decrease in zidovudine plasma exposure during pregnancy is not known. Like the other NRTIs, zidovudine is a prodrug that requires intracellular metabolism by cellular enzymes to the active triphosphorylated nucleotide form (102). The rate-limiting step in zidovudine activation is the conversion of zidovudine monophospate to diphosphate, catalyzed by cellular thymidylate kinase (112,113). This enzyme is saturated at relatively low substrate concentrations, so that intracellular concentrations of zidovudine monophosphate greatly exceed those of zidovudine di- and triphosphate, and plasma zidovudine concentrations do not correlate with intracellular concentrations of zidovudine triphosphate (112,113). The half-life of intracellular zidovudine triphosphate exceeds that of zidovudine in plasma, averaging 3 to 4 hours (114,115). As a result, zidovudine dose and plasma zidovudine concentration do not directly correlate with the concentration of intracellular phosphorylated metabolites or clinical effects (102,116). No studies of intracellular zidovudine metabolites have been performed in women receiving chronic oral dosing during pregnancy. The prenatal zidovudine-dosing regimen used in the PACTG 076 study was 100 mg given five times a day, which was the standard adult regimen at that time (6). With awareness of the importance and persistence of intracellular zidovudine triphosphate and the lack of correlation between plasma zidovudine concentrations and clinical effect, less frequent dosing regimens have been developed that appear to be equally effective and encourage adherence (2). The current standard adult zidovudine regimens of either 200 mg every 8 hours or 300 mg every 12 hours are generally used in pregnant women (13).
Zidovudine appears to cross the placenta well, with roughly equivalent concentrations in maternal plasma compared with amniotic fluid during pregnancy and in maternal plasma at the time of delivery compared with cord blood (110,111,117). Primate and human studies suggest that zidovudine moves across the placenta by simple diffusion (118,119,120,121). Regimens to prevent mother-to-child HIV transmission generally incorporate antiretroviral dosing during labor to ensure that suppression of HIV viral replication continues throughout labor and that protective plasma antiretroviral concentrations are present in the infant at the time of birth (13). The first intrapartum regimen studied was 140 mg of zidovudine given intravenously every 4 hours (109). This regimen led to average zidovudine concentrations of 1.15 μg per mL at the end of the infusion. However, zidovudine concentrations in umbilical cord and infant plasma at the time of delivery were highly variable and were dependent on the length of time separating birth and the last zidovudine dose (109). As a result, continuous intravenous infusion was investigated as a means of ensuring adequate zidovudine exposure at the time of birth. With continuous intravenous infusion of zidovudine using a 2 mg per kg loading dose followed by 1 mg per kg per hour, the average zidovudine plasma concentration at the time of birth was 0.82 μg per mL in the mother and 0.75 μg per mL in the newborn (109). Continuous intravenous zidovudine infusion during labor was part of the regimen used in the PACTG 076 protocol, the first study to demonstrate the efficacy of antiretrovirals in preventing mother-to-child HIV transmission, and remains part of standard clinical practice for HIV-infected pregnant women (6,13). Zidovudine is the only antiretroviral commercially available in a formulation for intravenous administration.
The human placenta phosphorylates zidovudine to its active metabolite, and the intracellular mechanisms needed to phosphorylate zidovudine are also present in the fetus (99). Intracellular concentrations of phosphorylated metabolites of zidovudine in maternal and cord blood have been studied following administration of continuous infusions during labor. Median levels of zidovudine monophosphate and triphosphate were similar in maternal (1,556 and 67 fmol per 106 cells) and cord (1,464 and 70 fmol per 106 cells) blood, but considerable variability was observed among study subjects (122). These values are two to three times higher than those reported in HIV-infected adults receiving oral zidovudine and, assuming uniform intracellular distribution of zidovudine triphosphate, around five times the zidovudine triphosphate 50% inhibitory concentration (IC50) for HIV reverse transcriptase (∼0.05 μM) (122). Although continuous intravenous infusion of zidovudine during labor provides maternal and cord blood intracellular zidovudine triphosphate levels consistent with high antiviral activity, the relative contributions of maternal, placental, and fetal zidovudine triphosphate in preventing intrapartum and early postpartum mother-to-child transmission are unknown.
Although the full zidovudine regimen used in the PACTG 076 protocol is standard of care in the United States and other developed countries, it is not available in resource poor areas of the world where the majority of HIV-infected women live. Less intensive zidovudine regimens that are more practical for use in these areas have been developed (14,123). These regimens start later in gestation, include oral dosing (rather than intravenous) during labor, and provide short or no postpartum newborn dosing. While these less intensive regimens have been shown to reduce mother-to-child HIV transmission, they are less effective than the full 076 protocol regimen (5). Zidovudine pharmacokinetics following oral dosing during labor has been described. In a study of five US women, trough plasma zidovudine concentrations following oral dosing with 300 mg every 3 hours
during labor ranged from 0.11 to 1.34 μg per mL (124). In a study conducted in Bangkok, median cord blood concentrations following zidovudine dosing with 300 mg every 3 hours during labor were 0.25 μg per mL, considerably less than with continuous intravenous infusion (125). Simulations based on the pharmacokinetic parameters observed in the US women suggest that a regimen consisting of an oral loading dose of 600 mg followed by 400 mg every 3 hours during labor would produce plasma concentrations equivalent to those seen with continuous infusion (124). Although this regimen has been used in clinical trials, no data are available describing plasma or intracellular zidovudine concentrations achieved with its use (20).
during labor ranged from 0.11 to 1.34 μg per mL (124). In a study conducted in Bangkok, median cord blood concentrations following zidovudine dosing with 300 mg every 3 hours during labor were 0.25 μg per mL, considerably less than with continuous intravenous infusion (125). Simulations based on the pharmacokinetic parameters observed in the US women suggest that a regimen consisting of an oral loading dose of 600 mg followed by 400 mg every 3 hours during labor would produce plasma concentrations equivalent to those seen with continuous infusion (124). Although this regimen has been used in clinical trials, no data are available describing plasma or intracellular zidovudine concentrations achieved with its use (20).
Zidovudine is rapidly cleared in adults by hepatic glucuronide conjugation, followed by renal excretion of mostly conjugated metabolite and some unchanged drug (104). Both hepatic glucuronidation and renal function are known to be depressed in infants immediately after birth, so not surprisingly, the washout t1/2 of transplacentally acquired zidovudine is extremely prolonged, averaging 13 hours (126). Zidovudine elimination increases rapidly during the first days of life, with t1/2 averaging 3 hours during days 3 to 10 (126). A population analysis combining zidovudine pharmacokinetic data from six studies demonstrated a further increase in zidovudine clearance over the first 2 months of life, with clearance reaching adult levels by 4 to 8 weeks of life (Fig. 22.2) (127). The developmental pattern of the increase in zidovudine clearance in the infant parallels that of bilirubin, whose primary route of elimination is also via hepatic glucuronidation (128). However, although bilirubin and zidovudine clearance mature in parallel, they are metabolized by different isoenzymes of the uridine diphosphate-glucuronosyltransferase (UGT) family, with zidovudine metabolized primarily by UGT 2B7 and bilirubin by UGT 1A1 (129,130). The current FDA-approved dosing recommendation for zidovudine in infants from birth to 3 months of age is 2 mg per kg PO or 1.5 mg IV every 6 hours. To facilitate adherence, many clinicians and some research protocols have used 4 mg per kg PO every 12 hours (17,131). Zidovudine clearance is further decreased in premature infants (Fig. 22.2) and a dosing reduction is needed to avoid the accumulation of potentially toxic serum zidovudine concentrations (132,133). Infants born before 35 weeks’ gestation who require zidovudine should receive initial doses of 2.0 mg per kg PO or 1.5 mg per kg IV every 12 hours. Zidovudine dosing frequency should increase to every 8 hours at 2 weeks of age if gestational age at birth is above 30 weeks or at 4 weeks of age if gestational age at birth is less than 30 weeks (134).
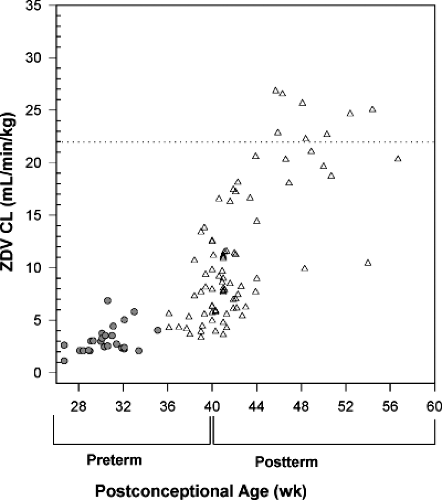 Figure 22.2. Zidovudine clearance plotted against postconceptional age (gestational age at birth plus postnatal age) for infants from birth to age 5 months. Open triangles, term infants; solid circles, preterm infants; broken line, average adult clearance [from Mirochnick et al. (127)]. |
Zidovudine is the drug with the longest history of use during pregnancy and the most safety information. High dose administration of zidovudine to female adult rodents is associated with the development of vaginal tumors (135). These tumors have not been reported in humans and are thought to result from chronic local exposure of the vaginal epithelium to high urine concentrations of unmetabolized zidovudine due to patterns of zidovudine metabolism and female anatomy unique to rodents. Bone marrow depression is a common toxicity of zidovudine, and mild, transient depression of hematologic parameters has been observed in the newborn after exposure to the full PACTG 076 regimen and to less intensive regimens (6,136). Hemoglobin was decreased by an average of 1 mg per dL at 3 weeks of age in newborns exposed to zidovudine in PACTG 076 compared with those exposed to placebo. By 12 weeks of age, no differences were seen in hemoglobin between the two groups (6). No adverse effects of zidovudine exposure have been detected during follow-up of the PACTG 076 infants followed for up to 5.6 years (48). Eighteen-month follow-up of a group of Thai infants randomized to exposure to a short-course prenatal zidovudine regimen or placebo similarly showed no adverse effects of zidovudine exposure (49). No evidence of cardiac toxicity could be found when comparing zidovudine exposed and unexposed infants born to HIV-infected mothers (137). Sufficient numbers of exposures to zidovudine in humans have been monitored in the Antiretroviral Pregnancy Registry to be able to determine that first trimester zidovudine exposure is not associated with a 1.5-fold or greater increase in the risk of overall birth defects or with a twofold or greater increase in the risk of cardiovascular or genitourinary system defects (138). However, the Women and Infants Transmission Study documented a 10-fold increased risk of hypospadias with first trimester zidovudine use (139). The potential relationship between perinatal zidovudine exposure and persistent infant mitochondrial toxicity has been discussed previously in this chapter.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


