Anticonvulsants
Victoria Tutag Lehr
Merene Mathew
Harry T. Chugani
Jacob V. Aranda
Epilepsy during infancy and childhood represents a therapeutic challenge because as many as 30% of children continue to have seizures refractory to treatment (1). Antiepileptic drugs (AEDs) remain the primary treatment modality for seizures (2,3,4,5). Multidrug regimens are often necessary to maximize seizure control and minimize adverse effects. During the past 20 years, numerous second- and third-generation AEDs have been approved in the United States (2,3,4,5). These AEDs include felbamate, gabapentin, lamotrigine, topiramate, tiagabine (TIG), levetiracetam (LEV), oxcarbazepine (OXC), zonisamide (ZNS), pregabalin, lacosamide, and rufinamide (2,3,5). Overall, these newer agents have shown more favorable side effect profiles, fewer drug–drug interactions, and less complex monitoring requirements compared with older AEDs (5,6,7). Pediatric clinicians (non-epileptologists) have been slow to replace older agents with new AEDs for many reasons, including long experience with older AEDs, insufficient postmarketing experience assessing relative efficacy, tolerability, and safety in infants and children, and increased relative cost (7). Many of the new AEDs are broad spectrum in mechanism of action offering improved seizure control with fewer adverse effects, particularly for those pediatric patients with intractable seizure disorders.
There is a paucity of data from well-designed clinical trials of the newer AEDs in infants and children. Extrapolation of data from trials in adults to pediatric patients is questionable, given age-related differences in pharmacokinetics, seizure types, and seizure etiology (8,9,10). In addition, children frequently have comorbidities such as mental retardation and behavioral disorders. Adverse effects and the variable pharmacokinetics of an AED, as a child undergoes maturational changes from birth to adolescence, with or without the presence of comorbidities, complicate anticonvulsant therapy (8,9,10,11,12,13). Pharmacokinetic differences may be partially responsible for age-related differences in the incidence of adverse effects observed with AEDs (12,13).
Interactions of AEDs with other agents are common; therefore, it is important to anticipate and minimize the risks of interactions to avoid loss of seizure control or development of toxicity (13,14,15). The newer AEDs such as lacosamide, LEV, and topiramate do not extensively induce metabolism of other AEDs, thereby offering an advantage over older agents (5,6,15). The risk–benefit ratio of AEDs must be considered when developing medication regimens.
In January 2008, the US Food and Drug Administration (FDA) issued an alert about a possible increased risk for suicidality associated with use of AEDs when prescribed for epilepsy (16,17,18,19). The relationship between suicidality and epilepsy is a complex, multifactorial issue (16). The contribution of AEDs to suicidality is unclear in view of confounders such as concomitant drug therapy, mental health diagnoses, effects of chronic epilepsy, and severity of epilepsy. Parents, caregivers, and patients must be reassured and counseled about the risks of withdrawing or discontinuing AED therapy without clinician supervision or approval. However, it is prudent that all patients with epilepsy be routinely evaluated for depression, anxiety, and suicidality, especially during periods of AED dosage adjustment (16,18,19).
Initiation of Anticonvulsant Therapy
Initial anticonvulsant therapy is selected on the basis of seizure type and findings on the electroencephalogram (20). Additional factors influencing the selection of AED therapy include patient age, gender, history of drug reactions, concomitant disease states and medications, ease of administration, cost, and clinician familiarity with the individual AED.
Phenytoin
Phenytoin continues to be used in pediatrics for status epilepticus, generalized tonic–clonic seizures, and partial seizures with or without secondary generalization (21,22). This hydantoin is often administered as a second-line agent for status epilepticus after administration of benzodiazepines have failed to control seizures (23).
Phenytoin continues to be considered second-line therapy for managing neonatal seizures after phenobarbital has failed (24,25,26,27). However, in a randomized, clinical trial by Painter et al. in 59 neonates with seizures, phenytoin and phenobarbital were equally, but incompletely, effective in controlling neonatal seizures (25). When either of these medications was administered as single therapy, fewer
than half of the babies had control of their seizures (26). Phenytoin and phenobarbital continue to be used in the management of neonatal seizures, although newer agents such as LEV are gaining popularity (27). In a large multicenter retrospective study (n = 6,099 infants) of neonatal seizure management, phenytoin was the second most frequently administered nonbenzodiazepine (27).
than half of the babies had control of their seizures (26). Phenytoin and phenobarbital continue to be used in the management of neonatal seizures, although newer agents such as LEV are gaining popularity (27). In a large multicenter retrospective study (n = 6,099 infants) of neonatal seizure management, phenytoin was the second most frequently administered nonbenzodiazepine (27).
Phenytoin’s primary anticonvulsant effect at therapeutic concentrations is related to use-dependent blockade of voltage-sensitive sodium channels, thus inhibiting repetitive neuronal firing (4). Additional actions include alteration of Na+, K+, and Ca2+ conduction; membrane potentials; and concentrations of amino acids, norepinephrine, acetylcholine, and γ-aminobutyric acid (GABA).
The major route of phenytoin metabolism is hepatic oxidation by cytochrome P450 (CYP450) enzymes CYP2C9 and CYP2C19 to the inactive metabolite 5-(p-hydroxyphenyl)-5-phenylhydantoin (28). This metabolic pathway is capacity limited and therefore saturable. As phenytoin serum concentrations increase, the fraction of drug eliminated per unit time decreases. Small increases in dose can result in disproportionately large increases in phenytoin serum concentration. These nonlinear kinetics occur in adults as well as in children of all ages and is described by Michaelis–Menten equations (29). Therefore, smaller incremental dose increases are recommended, as therapeutic phenytoin serum concentrations are approached.
Initial phenytoin dosing for neonates is 15 to 20 mg per kg intravenous or oral as a loading dose, followed by 4 to 7 mg per kg per day in divided doses every 12 hours as a maintenance dose. To avoid cardiac toxicity, bradyarrhythmias, and hypotension associated with the propylene glycol and ethanol constituents of parenteral formulations, the maximum rate of phenytoin intravenous infusion is 0.5 mg per kg per minute. Phenytoin injection is compatible only with normal saline. Oral phenytoin loading doses need to be administered in two to three divided doses and administered every 2 hours to optimize absorption. The maintenance dose is initiated 12 hours after administration of the loading dose (30).
Phenytoin dosage must be slowly adjusted according to the individual patient’s requirements, clinical response, and serum concentrations (30). Because of nonlinear kinetics, it is prudent to initiate therapy with the lower end of the dosing range and increase incrementally until clinical response, assessing for toxicity. Initial maintenance dosages are based on age and weight: neonates (<4 weeks), 3 to 5 mg per kg per day; infants (4 weeks to <1 year), 4 to 8 mg per kg per day; children (1 to <12 years), 4 to 10 mg per kg per day; adolescents (12 to <18 years), 4 to 8 mg per kg per day; adults (18 years and older), 4 to 7 mg per kg per day.
Caution is advised when changing dosage forms of phenytoin, as these vary in phenytoin content. For example, the parenteral form, phenytoin sodium, contains 92% phenytoin and the oral suspension is free phenytoin acid. As the phenytoin dose is increased and the therapeutic range is approached, the difference in phenytoin content between the dose formulations becomes critical to avoid toxicity secondary to a disproportionate increase in serum concentration (29).
Phenytoin has a narrow therapeutic range for the general population of 10 to 20 μg per mL for total drug concentration and 1 to 2 μg per mL for unbound or free drug (31). Free phenytoin serum determinations are essential in the setting of hypoalbuminemia, azotemia, or reduced binding because the free fraction of the drug determines therapeutic effect and toxicity. Secondary to reduced plasma protein binding and decreased metabolic capacity, the therapeutic range is lower during the neonatal period, 8 to 15 μg per mL (32). Phenytoin binding at approximately 3 months of age resembles that of adults.
Infants exposed to phenytoin in utero or to phenobarbital therapy prior to phenytoin will have increased phenytoin elimination secondary to hepatic enzyme induction (33). There is considerable variability in correlation of clinical response with serum concentration in cases of decreased plasma protein binding, hypoalbuminemia, uremia, renal dysfunction, and hyperbilirubinemia. During the first 2 weeks of life, the infant’s metabolic capacity increases significantly, resulting in increased phenytoin dose requirements. In general, infants have the highest phenytoin metabolic capacity, which generally results in dose requirements up to four times that of adults. During childhood, phenytoin metabolism decreases and approaches that of adults at around 10 years of age (29) (Fig. 37.1).
Therapeutic drug monitoring (TDM) is recommended for phenytoin to optimize therapeutic effect and minimize risk of toxicity. Frequency of serum sampling for phenytoin depends on the clinical situation. After administration of a phenytoin-loading dose, serum sampling is often performed to ascertain achievement of therapeutic serum concentrations. Samples must not be drawn within 1-hour postend phenytoin infusion to allow for drug distribution.
For oral and intravenous maintenance therapy, trough serum concentrations are usually drawn just prior to the morning dose. On initiating a maintenance regimen, phenytoin concentrations are drawn within 3 to 4 days starting therapy. This is not a steady-state determination; however, subtherapeutic or supratherapeutic concentrations may be detected at this time, thus avoiding breakthrough seizures or toxicity. Once a stable phenytoin regimen is achieved, serum sampling is performed every 1 to 2 weeks in hospital or every 1 to 6 months in outpatients. Sampling is indicated when a change in clinical status occurs, when a drug with potential for interacting with phenytoin is added to or deleted from the regimen, when changing phenytoin dosage forms or dosage, or to verify absorption of the dose or compliance. Significant variations in the pharmacokinetics occur during the neonatal period with intravenous and oral phenytoin therapy (34,35). Controversy exists regarding whether neonates have the ability to absorb oral formulations of phenytoin (36). However, oral phenytoin therapy has been documented to achieve predicted serum concentrations in a study of premature infants (25). In addition to the rapidly changing elimination kinetics, concomitant therapy with potent enzyme inducers, such as phenobarbital and carbamazepine (CBZ), may result in unpredictable fluctuations in phenytoin serum concentration (28).
Numerous clinically significant pharmacokinetic and pharmacodynamic drug interactions involving phenytoin have been described (6,14,15,37,38). Agents with potential for interacting with phenytoin are too numerous to list here as new drugs are developed and practice evolves. Clinicians
are encouraged to consult with pharmacists and to review the current drug interaction literature when prescribing phenytoin therapy for their patients. Phenytoin serum concentrations may be increased by zidovudine and reduced by continuous nasogastric feedings (37). Concomitant therapy with valproic acid (VPA) significantly increases the free fraction of phenytoin. Interactions with other AEDs and phenytoin are prevalent and may be significant (14,15).
are encouraged to consult with pharmacists and to review the current drug interaction literature when prescribing phenytoin therapy for their patients. Phenytoin serum concentrations may be increased by zidovudine and reduced by continuous nasogastric feedings (37). Concomitant therapy with valproic acid (VPA) significantly increases the free fraction of phenytoin. Interactions with other AEDs and phenytoin are prevalent and may be significant (14,15).
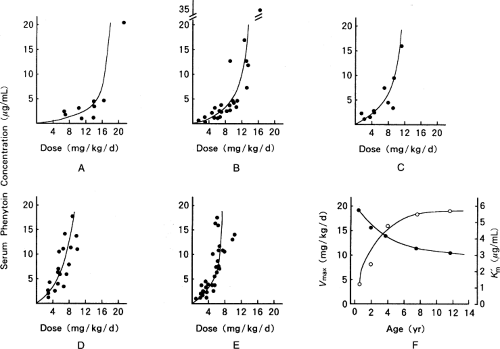 Figure 37.1. (A–E) Phenytoin dose and serum concentration in different age groups. (F) Change of Vmax (•) and K′m (o) with increasing age. (Modified from Nishihara K, Kohda Y, Saitoh Y, et al., lgaku-No-Ayumki 1978;107:512–515, with permission.) |
Phenytoin may decrease serum concentrations and the effectiveness of lamotrigine, VPA, felbamate, ethosuximide, primidone, potentially resulting in loss of seizure control. A consideration for adolescents receiving phenytoin therapy with oral contraceptives is the potential for phenytoin to lower estrogen and progestin serum concentrations, resulting in contraceptive failure. Higher dose oral contraceptives in addition to barrier methods are recommended for these patients (6,38). The potential for precipitating drug interactions must be evaluated whenever another agent is added to or removed from a therapeutic regimen containing phenytoin.
Fosphenytoin
Fosphenytoin sodium (Cerebryx), 5,5-diphenyl-3-(phosphonooxy) methyl-2,4-imidazolidine-dione disodium, is the phosphorylated prodrug of phenytoin (39). Fosphenytoin is supplied as a 75-mg per mL parenteral injection, which is equivalent to 50 mg phenytoin sodium per mL. Fosphenytoin must be prescribed in terms of phenytoin equivalents (PEs), with 1 mg PE equal to 1.5 mg fosphenytoin. Phenytoin toxicity may result from misinterpretation of fosphenytoin dosage. Advantages over phenytoin include aqueous solution without propylene glycol and a more neutral pH of 8.6. Fosphenytoin is compatible with dextrose 5% water or normal saline, and also may be administered via intramuscular injection. Cardiac toxicity during intravenous infusion and local reactions at the site of injection occur less frequently with fosphenytoin than with phenytoin. Transient parethesias, burning, and pruritis have occurred during intravenous infusion of fosphenytoin. The rate of intravenous infusion for fosphenytoin is three times faster than that of phenytoin infusion, providing an advantage for rapid loading in status epilepticus.
Fosphenytoin is cleaved to phenytoin by nonspecific phosphatases in red blood cells and in hepatic and other tissues. Conversion half-life is 8 to 15 minutes and may be prolonged in renal or hepatic insufficiency (40). Fosphenytoin displaces phenytoin from plasma proteins, resulting in increased free phenytoin concentrations until the conversion is complete. Phenytoin serum concentrations should not be drawn until the conversion to phenytoin is complete, approximately 2 hours post-end infusion of fosphenytoin or 4 hours after intramuscular administration. It has been postulated that the increased free fraction may be cleared rapidly in infants, resulting in difficulty in maintaining therapeutic serum concentrations.
Fosphenytoin is approximately 10 times the cost of phenytoin injection; therefore, cost–benefit analysis is indicated. Restricting fosphenytoin use to patients without intravenous access or with prior local reactions to phenytoin administration, or for cases of status epilepticus, has been recommended (41,42). There is no apparent therapeutic benefit in using fosphenytoin over phenytoin that justifies the higher cost in other situations (43).
There are limited data on the use of fosphenytoin in the newborn (44,45,46). Fosphenytoin was administered to two extremely low-birth-weight infants with apparent adequate conversion to phenytoin and no adverse effects (44). Takeoka et al. reported seizure control in four infants ranging in age from 34 weeks to 1 year treated with fosphenytoin; however, doses of up to 10 mg PE per kg per day were required to maintain therapeutic phenytoin serum concentrations (45). Three of the infants had been receiving phenobarbital therapy at the time of fosphenytoin administration. In addition to probable induction of CYP450 by phenobarbital, the investigators suggested that the infants may have had an enhanced phenytoin rate of elimination. There was no significant difference in conversion rate of fosphenytoin to phenytoin in two multicenter studies of 78 patients ranging in age from 1 day to 16 years. Subjects received fosphenytoin loading doses of 18 to 20 mg PE per kg per minute via intravenous infusion (62/78) or 12 to 20 mg PE per kg per minute via intramuscular injection (16/78). Mean fosphenytoin-to-phenytoin conversion half-life was 8.3 minutes (2.5 to 18.5 minutes), which is similar to adult values (7.9 minutes). There was no significant difference in conversion half-life across age groups. Total and free phenytoin serum concentrations were similar to those in adults. There were no documented cases of toxicity or serious adverse effects. Infants and children in status epilepticus are at high risk for site reactions from phenytoin intravenous infusions. Fosphenytoin provides an effective, well-tolerated alternative to phenytoin in infants and children where venous access is not available and/or rapid loading is desirable (46).
Phenobarbital
Phenobarbital, the 5-ethyl-5-phenyl-substituted barbiturate, remains first-line management for neonatal seizures (24,27,47). However, it is frequently ineffective in achieving complete seizure control in neonatal seizures (47). A retrospective study of infants with neonatal seizures (n = 146) showed no benefit of phenobarbital treatment after discharge home with respect to preventing seizure recurrence or long-term disability compared with no treatment in a multicenter retrospective study (48). Although clinical trials are needed, newer agents such as LEV and topiramate may develop a role in the management of neonatal seizures as experience is gained with their use in the NICU population (49,50).
Other indications are generalized tonic–clonic seizures, partial seizures, and prolonged febrile convulsions. Advantages of phenobarbital include a wide spectrum of seizure activity, wide therapeutic range, availability of parenteral and oral dose forms, low cost, and extensive experience of use in pediatrics. Disadvantages include respiratory depression, sedation, physical dependence, negative cognitive effects, hyperactivity, and potential adverse effects on developing neuronal cells (51,52).
The anticonvulsant effect of phenobarbital is related to potentiation of inhibitory neurotransmission by prolonging the open state of GABA-mediated sodium channels. Glutamate-induced excitatory transmission is decreased and neurotransmitter release from nerve terminals is diminished via blocking of L-type and N-type calcium currents. Selective suppression of abnormal neurons may also contribute to its therapeutic effect (52).
Phenobarbital has a large volume of distribution, distributing into all tissues, with approximately 50% bound to plasma protein. Volume of distribution decreases with increasing gestational age as total body water decreases and body fat increases. These changes in volume of distribution may result in high (interpatient) variability in phenobarbital serum concentrations achieved after standard loading doses. In general, initial dosing recommendations for 10 to 20 mg per kg assume a volume of distribution of 1.0 L per kg to achieve serum concentrations of 15 to 20 mg per mL. Therapeutic phenobarbital serum concentrations are 10 to 30 mg per mL. Reduced binding on the order of 20% to 25% has been demonstrated in neonates (53,54). Because phenobarbital is a weak acid, lower serum pH will enhance tissue penetration. Distribution across the blood–brain barrier is relatively slow (15 to 20 minutes after peak serum concentrations); therefore, dosing protocols need to allow for equilibration after administration. Wide interindividual variability in volume of distribution and elimination among neonates makes it necessary to measure phenobarbital serum concentrations (54,55).
The primary route of elimination for phenobarbital is metabolism via hepatic microsomal CYP450 enzymes and NADPH–cytochrome c reductase. Dosing reduction is advised in hepatic insufficiency. Phenobarbital clearance does not appear to undergo significant changes due to autoinduction, although it is a potent inducer of other hepatically metabolized agents such as theophylline, CBZ, phenytoin, cimetidine, and digoxin (14). Serum concentrations of phenobarbital may be increased by VPA or, in some instances, phenytoin. Serum concentration monitoring of phenobarbital is indicated when potentially interacting agents are added to or removed from therapeutic regimens. Clinicians are encouraged to consult with pharmacists and review the current literature and labeling when prescribing medications for patients taking phenobarbital to avoid precipitating a drug or drug–disease state interaction.
Phenobarbital has a long elimination half-life, with newborns demonstrating the slowest clearance (average 100 to 200 hours). Premature neonates may have unexpectedly increased clearance of phenobarbital, possibly secondary to increased liver size compared to body weight. Touw et al. reported that total body clearance of phenobarbital per kilogram of body weight tends to increase with increasing fetal maturity (55). Therefore, extremely premature infants would have not yet undergone the decrease in liver size relative to increasing body weight that occurs during the final weeks of gestation. During the first weeks of life, considerable variability occurs in phenobarbital elimination as hepatic enzymes mature or as a result of enzyme reduction from exposure to other drugs (53,54). Phenobarbital clearance
rapidly increases during the first 2 weeks of life, peaking between 6 months and 12 months of age.
rapidly increases during the first 2 weeks of life, peaking between 6 months and 12 months of age.
The loading dose of phenobarbital for the management of neonatal seizures is 20 mg per kg intravenous (24). The goal is to achieve a phenobarbital serum concentration of 15 to 40 μg per mL. Serum concentrations above 40 to 50 μg per mL may produce respiratory depression and coma, with 80 μg per mL associated with respiratory depression and death. There are reports of neonates tolerating phenobarbital serum concentrations of 60 to 80 μg per mL with respiratory support; however, bradycardia is frequently associated with serum concentrations greater than 50 μg per mL.
Additional doses of 5 to 10 mg per kg administered at 30-minute to 1-hour intervals may be required if seizures persist. Maintenance doses of 6 mg per kg per day are administered every 12 hours in divided doses. The long half-life of phenobarbital precludes the need for continuous infusions. However, continuous administration of daily doses of 5 mg per kg per day or more may result in accumulation of phenobarbital during the first weeks of life; therefore, doses of 2 to 4 mg per kg per day may be more appropriate for neonates younger than 2 weeks.
Studies have demonstrated that infants may require up to 40 mg per kg total loading dose (53,55). Effective seizure control has been related to phenobarbital dose, with 70% control with doses of 40 mg per kg (24). Neonates receiving extracorporeal membrane oxygenation may require larger phenobarbital doses to achieve effective serum concentrations secondary to a larger volume of distribution (56). Older studies report efficacy rates between 32% and 36% with standard dosing (57,58,59).
Children 1 to 18 years of age usually require phenobarbital loading doses of 10 to 20 mg per kg. The rate of administration is 2 mg per kg per minute for children weighing less than 40 kg, and not more than 100 mg per minute for children weighing more than 40 kg; the rate is 60 mg per minute for adults. Maintenance doses are 3 to 5 mg per kg per day for children 1 to 15 years of age, and 2 mg per kg per day for adults.
An initial phenobarbital serum concentration may be drawn 2 to 3 hours after administration of a loading dose to verify serum concentration (60,61,62). Secondary to a long half-life, steady-state serum concentrations may not be achieved for 2 to 4 weeks in neonates and infants. Serum concentration sampling may be repeated after 3 to 4 days of maintenance dosing to determine if dose titration is necessary; however, this does not reflect the steady-state phenobarbital serum concentration. Documentation of steady-state serum concentration should be performed after 3 to 4 weeks of therapy. Indications for phenobarbital serum concentration sampling include loss of seizure control, possible toxicity, dosage changes, and addition or deletion of potentially interacting agents.
Carbamazepine
Carbamazepine, CBZ, an iminodibenzyl derivative (iminostilbene) with a tricyclic antidepressant structure, has been used in adults and children as anticonvulsant for older than 30 years (63,64,65). This drug remains one of first-line treatments for partial motor, partial complex, and secondarily generalized tonic–clonic seizures. CBZ is not recommended for first-line management of primary generalized seizures due to possible exacerbation of these seizures (66). There is increasing use of CBZ for management of neonatal seizures (67,68,69). Advantages over other older AEDs include less sedation and a mood stabilization effect (70,71). Serious, although rare, aplastic anemia and agranulocytosis have been associated with CBZ therapy (72). Leukopenia and thrombocytopenia may occur, requiring hematologic monitoring (73). Rash, hypersensitivity reactions, including Stevens–Johnson syndrome, and toxic epidermal necrolysis are possible (74). CBZ has also been associated with significant weight gain (75).
Carbamazepine is available as a liquid suspension (Tegretol, 50 mg per mL), chewable tablets (Tegretol, 100 mg per mL), regular tablets (Tegretol and generic, 200 mg), sustained-release granules (Carbitrol, 200, 400 mg), and controlled-release formulation (Tegretol XR, 100, 200, 400 mg). There is no parenteral dosage form of CBZ. There may be considerable variability in rate of absorption of generic CBZ formulations compared with Tegretol, and therefore changing formulations must be performed with caution and close monitoring (76).
Carbamazepine’s mechanism of action is similar to that of phenytoin, that is, use-dependent blockade of voltage-sensitive sodium channels, resulting in neuronal membrane stabilization and inhibition of repetitive firing of neurons (63). Other anticonvulsant effects include presynaptic decrease of synaptic transmission and possibly potentiation of postsynaptic effects of GABA.
The primary elimination pathway of CBZ is metabolism via the CYP3A4 and CYP1A2/2C8 isozymes (75). Half-lives of CBZ in adults are 12 to 17 hours, with shorter values in children and infants. Neonates exposed to CBZ in utero have been reported to show half-lives of 8.2 to 48.0 hours. An active metabolite, CBZ-10,11-epoxide, is formed in a 0.1:0.2 ratio, and is inactivated via epoxide hydrolysis. CBZ induces its own metabolism (autoinduction) in a dose-dependent manner, resulting in decreased half-life and increased dose requirements several weeks after initiating or adjusting dosing. As a result, CBZ is prone to interactions with other drugs metabolized by the CYP450 enzyme system (77,78,79). Common drug interactions between CBZ and drugs commonly prescribed in the newborn include phenytoin (variable effect, phenytoin serum concentrations may increase, decrease, or remain unchanged, or CBZ concentrations may decrease) and phenobarbital (decreased CBZ serum concentrations) (6,14,77). As CBZ is approximately 75% bound to albumin (and CBZ-10,11-epoxide is approximately 50% albumin bound), there is potential for drug interactions that involve binding (14,38,78). Valproate will increase the unbound fraction of CBZ-10,11-epoxide, possibly resulting in increased neurotoxicity; however, for many patients, this interaction may not be clinically significant. Concomitant therapy with LEV may potentiate adverse central nervous system (CNS) effects of CBZ (79).
Carbamazepine has been used for controlling neonatal seizures as primary or second- or third-line therapy after phenobarbital and phenytoin (67,68,69). Loading doses of 10 mg per kg via nasogastric tube followed by maintenance doses of 7 to 23 mg per kg daily in two to three divided doses have been reported to be safe and effective in neonates,
including preterm infants (gestational age <30 weeks, weight <1,000 g). Apparent adequate absorption of CBZ occurs even in the smallest, critically ill neonates, as therapeutic CBZ serum concentrations were achieved in these studies. Further investigation is needed to determine the safety and efficacy of CBZ in neonates, as this agent offers another therapeutic option for management of neonatal seizures. CBZ requires serum concentration monitoring for optimal therapeutic benefit in view of interpatient variability in pharmacokinetics and narrow therapeutic range (80,81).
including preterm infants (gestational age <30 weeks, weight <1,000 g). Apparent adequate absorption of CBZ occurs even in the smallest, critically ill neonates, as therapeutic CBZ serum concentrations were achieved in these studies. Further investigation is needed to determine the safety and efficacy of CBZ in neonates, as this agent offers another therapeutic option for management of neonatal seizures. CBZ requires serum concentration monitoring for optimal therapeutic benefit in view of interpatient variability in pharmacokinetics and narrow therapeutic range (80,81).
Oxcarbazepine
Oxcarbazepine (Trileptal) is the 10-ketocogener of CBZ and requires hepatic metabolism to 10-monohydroxy-carbazepine, which is primarily responsible for its pharmacologic effects (82,83,84). Monohydroxy-CBZ is eliminated 96% by the kidneys. There is a lower potential for drug interactions due to reduced plasma protein binding and less potent induction of hepatic enzymes (83).
Oxcarbazepine was approved for use in the United States in 2000, but this agent has been used in Europe for several years (82,85). A recent review of European practice reveals OXC initial monotherapy as a treatment of choice for complex partial seizures in children (64). Indications for treatment of partial seizures as monotherapy in children aged 4 years or older through adulthood or as add-on therapy in partial seizures with or without secondary generalization for children 2 years of age through adulthood with partial seizures (85). Dose forms are an oral suspension, Trileptal, 300 mg per 5 mL (of OXC), and 150-mg, 300-mg, and 600-mg tablets. The oral tablets and suspension are interchangeable on a milligram-to-milligram equivalent basis. This is an important consideration when converting from one dosage form to another as a child matures and the tablet form may be preferred.
Oxcarbazepine has been referred to as a “cleaner” version of CBZ due to lack of autoinduction and less fewer CNS and hematologic adverse effects (82). However, there is an increased potential for hyponatremia with OXC compared with CBZ. This may be significant, as a serum sodium of less than 125 mEq per L has been reported in 2.5% of patients receiving OXC. Skin rashes and serious dermatologic manifestations such as Steven–Johnson syndrome and toxic necrolysis have developed during therapy with OXC (84). Cross reactivity with CBZ is possible; therefore, switching between these agents requires close monitoring in the event of a potential adverse reaction (74). As an inducer of hepatic microsomal enzymes, OXC is prone to drug interactions, although to a lesser extent than CBZ. OXC may inhibit hepatic metabolism of phenytoin and phenobarbital resulting potentially supratherapeutic concentrations of these agents. When adding or removing OXC from a patient’s drug regimen, close monitoring for potential drug–drug interactions is advised (6,14,15,38).
In a blinded, randomized, parallel-group study of infants and young children (1 month to <4 years of age, n = 128), with inadequately controlled partial seizures, OXC oral suspension was administered in low dose (10 mg per kg per d) or high dose (60 mg per kg per d) regimens as add-on therapy (86). Overall, high-dose OXC was significantly (p < .05) more effective than low dose in controlling partial seizures as measured by during continuous video-EEG monitoring. Adverse effects associated with OXC study treatment (>10% patients) were primarily mild somnolence and temperature elevation.
Valproic Acid and Sodium Valproate
Valproic acid (2-propylvaleric dipropyl acetic acid), the free acid form of sodium valproate, has been approved for anticonvulsant use in the United States for over 30 years (89). This agent is available as oral and parenteral formulations. The active form in plasma is the valproate ion; therefore, all dose forms in this discussion will be referred to as VPA.
Dose forms are valproate sodium for intravenous injection, 100 mg (of VPA) per mL (Depacon), and valproate sodium oral solution (Depakene) 250 mg per 5 mL. Other formulations are VPA liquid-filled capsules (Depakene) 250 mg; divalproex sodium capsules containing coated particles to mix with food (Depakote Sprinkle) 125 mg VPA equivalent; enteric-coated delayed-release tablets (Depakote) containing 125-, 250-, and 500-mg VPA equivalent; and extended-release tablets (Depakote ER) containing 250- and 500-mg VPA equivalent. The rate of gastric absorption of VPA is dose-form dependent; overall bioavailability is 80% to 90%. Food slows down the rate but not the extent of absorption. Peak serum VPA concentrations are achieved approximately 2 hours after oral administration of syrup or uncoated tablets, 3 to 5 hours after single-dose administration of the enteric-coated divalproex sodium tablet, and 7 to 13 hours after multiple dosing. Parenteral valproate sodium is indicated for situations when therapeutic serum concentrations need to be achieved rapidly, such as in status epilepticus, neonatal seizures, or following surgery when the patient cannot be given oral medications.
Valproic acid is effective for many seizure types including myoclonic, tonic, atonic, absence, generalized tonic–clonic seizures, and partial-onset seizures (89,90). Many clinicians consider this agent to be the drug of choice for absence or atypical absence seizures. VPA has been used in the management of neonatal seizures, usually as a second- or third-line alternative after failure with phenobarbital and phenytoin (91,92,93). There is some evidence that VPA may be effective in the management of Lennox–Gastaut syndrome and infantile spasms (IS) (94,95,96,97). VPA therapy is a strategy for the management of IS resistant to treatment with adrenocorticotropic hormone (ACTH), vigabatrin (VGB), or prednisone (97).
The broad spectrum of anticonvulsant activity of VPA has not been fully elucidated. Mechanisms of action include (a) use-dependent blockade of voltage-sensitive sodium channels, resulting in neuronal membrane stabilization and inhibition of repetitive firing of neurons; (b) increased brain concentrations of GABA possibly secondary to increased synthesis via glutamic acid decarboxylase;
(c) increasing GABA concentrations by inhibition of GABA transporter GAT-1, therefore blocking conversion of GABA to succinic semialdehyde; and (d) increased membrane potassium conduction (89).
(c) increasing GABA concentrations by inhibition of GABA transporter GAT-1, therefore blocking conversion of GABA to succinic semialdehyde; and (d) increased membrane potassium conduction (89).
Valproic acid is highly plasma protein bound in a concentration-dependent manner, with the free fraction increasing with increasing serum concentration. Binding is reduced in renal or hepatic disease, in uremia, or in the presence of other highly plasma-protein-bound drugs.
Valproic acid undergoes hepatic biotransformation via glucuronidation, beta and omega oxidation, hydroxylation, ketone formation, and desaturation (90). Glucuronidation with urinary excretion of the β-glucuronidation is the most important pathway. There are two metabolites with anticonvulsant activity, 2-ene VPA and 4-ene VPA. Hepatotoxicity and embryotoxicity are most likely associated with the 4-ene VPA metabolite. This serious adverse effect of VPA is more frequent in children younger than 2 years and anticonvulsant polytherapy and received a black box warning in the United States. Side effects frequently encountered during VPA therapy include weight gain, nausea, easy bruising secondary to thrombocytopenia, and tremor. Weight gain secondary to VPA therapy may be a major concern among clinicians, parents, and patients. A recent retrospective review of 94 patients aged 2 to 20 years taking VPA suggests that children may be less likely to gain significant amounts of weight while taking this medication compared with adult patients (98). These results must be documented in larger, prospective trials. The longer-acting VPA preparations have been reported to be associated with less weight gain when administered once daily (99).
The metabolism of VPA is sensitive to enzyme induction and inhibits the metabolism of other drugs (6,14,15,38). Therefore, patients receiving phenobarbital, CBZ, or primidone may have increased VPA dose requirements (100). Doses of phenobarbital or primidone may need to be reduced by 20% to 40% when VPA is added to a regimen containing these agents. VPA also inhibits the metabolism of ethosuximide and lamotrigine. Phenytoin serum concentrations may be decreased by the addition of VPA, or free fractions of phenytoin may be increased by binding displacement as described previously. It is prudent to monitor serum concentrations of concomitantly administered anticonvulsants as well as clinical signs and symptoms of toxicity whenever VPA therapy is initiated or discontinued.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


