Chapter 515 Anatomic Abnormalities Associated with Hematuria
515.1 Congenital Anomalies
Craig C. Porter and Ellis D. Avner
Gross or microscopic hematuria may be associated with many types of different malformations of the urinary tract. The sudden onset of gross hematuria after minor trauma to the flank is often associated with ureteropelvic junction obstruction or cystic kidneys (see Chapter 531).
515.2 Autosomal Recessive Polycystic Kidney Disease
Diagnosis
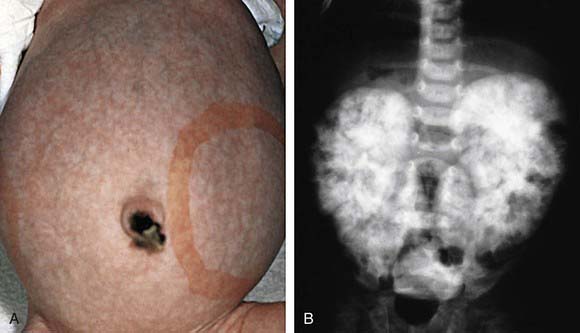
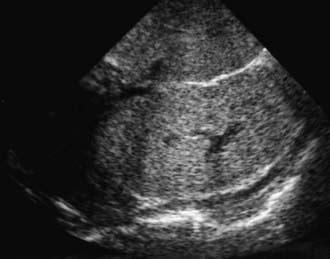
The diagnosis of ARPKD is strongly suggested by bilateral palpable flank masses in an infant with pulmonary hypoplasia, oligohydramnios, and hypertension and the absence of renal cysts by sonography of the parents (Fig. 515-1). Markedly enlarged and uniformly hyperechogenic kidneys with poor corticomedullary differentiation are commonly seen on ultrasonography (Fig. 515-2). The diagnosis is supported by clinical and laboratory signs of hepatic fibrosis, pathologic findings of ductal plate abnormalities seen on liver biopsy, anatomic and pathologic proof of ARPKD in a sibling, or parental consanguinity. The differential diagnosis includes other causes of bilateral renal enlargement and/or cysts, such as multicystic dysplasia, hydronephrosis, Wilms tumor, and bilateral renal vein thrombosis (Table 515-1). Prenatal diagnostic testing using genetic linkage analysis or direct mutation analysis is available in families with ≥1 affected child.