Analgesic Agents
Sinno H.P. Simons
Brian J. Anderson
Dick Tibboel
Introduction
It was generally believed that neonates were unable to experience pain (1). The importance of antinociceptive therapy in children, infants, and newborns has been increasingly acknowledged over the past two decades, leading to a burst of research and increased use of analgesic agents in this population. Research has concentrated on the development of pain assessment instruments and clinical trials investigating the effectiveness and safety of analgesics in infants. Many instruments containing behavioral and physiologic items have proved to be useful and reliable measures of postoperative and procedural pain in different age groups and to a lesser extent for individuals with mental handicaps. Knowledge about analgesic effects has been enlarged using pain assessment tools in randomized trials comparing different dose regimen and different agents (2). These trials have gained more insight into the specific pharmacokinetics (PK) and pharmacodynamics (PD) of analgesics in infants (3,4). PK and PD knowledge has increased over the past few years, but more research is necessary (5).
Pediatric pharmacokinetics is altered with age through the maturation of enzyme systems and physiologic processes responsible for absorption and elimination (6,7,8). Changing body composition alters disposition. Pharmacodynamic changes with age are poorly documented, although out of infancy they are commonly similar to adults (9). Purported pharmacodynamic differences between children and adults for drugs such as morphine, for example, disappear once PK differences are accounted for (10,11).
Analgesics in pediatric patients can be broadly divided into opioids and nonopioids.
Opioids
Opioid analgesics include naturally occurring agents (opium alkaloids) and synthetic opioid agonists that elicit morphine-like activity. The analgesic effects of opioids occur by activation of μ-, κ-, and/or δ-receptors in the central nervous system (CNS) (12). Each class of receptors is divided into subtypes that have different clinical effects. Analgesia is obtained by spinal or supraspinal activation of opioid receptors, leading to decreased neurotransmitter release from nociceptive neurons, inhibiting the ascending neuronal pain pathways and altering perception and response to pain (13). Opioid receptors also exist outside the CNS in the dorsal root ganglia and on peripheral terminals of primary afferent neurons (14).
The World Health Organization (WHO) Analgesic Ladder is a generally accepted guideline for the supply of analgesics; it was originally developed for the treatment of cancer pain. Mild pain should be treated with nonopioid analgesics [acetaminophen or nonsteroidal anti-inflammatory drugs (NSAIDs)], moderate pain should be treated with “weaker” opioids or combination products, and severe pain should be treated with stronger opioids (World Health Organization; Cancer Pain Relief; Albany NY: WHO Publications Center, 1986) (15). Opioids in children, infants, and newborns are reserved for moderate-to-severe types of pain, such as postoperative pain, sickle cell disease, and palliative care (16), or as an additive to acetaminophen or NSAIDs if pain is moderate. Furthermore, opioids may be used in the intensive care unit for pain or stress related to artificial ventilation, surgical procedures [chest tube placement, vessel canulation for extracorporeal membrane oxygenation (ECMO)], or painful conditions such as necrotizing enterocolitis. The most frequently used opioids are fentanyl and morphine, but codeine, oxycodone, methadone, hydromorphone, and meperidine are used in children, as well as fentanyl derivatives such as alfentanil and sufentanil. Recommended starting doses are shown in Table 46.1. All doses should, however, be adjusted to clinical circumstances and titrated to the individual patients’ needs. Doses aim to achieve a target concentration, but the correlations between the analgesic plasma concentrations and validated pain scores are weak (17,18).
Opioids produce adverse effects that may be minimized by appropriate drug selection and dosing. Respiratory depression, hypotension, glottic and chest wall rigidity, constipation, urinary retention, seizures, sedation, and bradycardia are well described. Continuous monitoring and frequent assessment of vital signs should be performed during opioid administration. Naloxone is a competitive opioid receptor agonist that reverses many of these side effects in appropriate dosage. Naloxone also antagonizes endorphin effects, and some morphine side effects can be
managed alternatively (e.g., neuromuscular blocking drugs for fentanyl muscle rigidity).
managed alternatively (e.g., neuromuscular blocking drugs for fentanyl muscle rigidity).
Table 46.1 Opioid Analgesics: Recommended Starting Doses in Neonates and Childrena | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 46.2 Onset, Peak, and Duration of Effects as well as Lipid Solubility of Opioids | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
Morphine
Mechanism of Action/Metabolism
The most common opioid used for pediatric pain is morphine. Morphine is a member of the morphinan-framed alkaloids. The drug is soluble in water, but lipid solubility is poor compared with that of other opioids (Table 46.2). Although morphine may also act on κ-opioid receptor subtypes (19), the analgesic effect of morphine is mainly caused by an activation of μ-receptors, as confirmed by a lack of analgesic effect of morphine in murine studies using μ-receptor knockout mice (20,21,22). Alterations of the morphine molecular structure change the pharmacologic activity and may have important clinical consequences (Fig. 46.1). The most important positions on the morphine molecule, next to the nitrogen atom (probably responsible for the analgesic activity, as modifications reduce penetration into the CNS), are the phenolic hydroxyl at position 3 and the alcoholic hydroxyl at position 6. Morphine is mainly metabolized by the enzyme UDP-glucuronosyl transferase 2B7 (UGT2B7) into morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) (23). Contributions to both the desired effect (analgesia) and the undesired effects (nausea, respiratory depression) of M6G are the subject of clinical controversy (24,25,26,27,28,29,30). M3G has been suggested to antagonize the antinociceptive and respiratory depressive effects of morphine and M6G (31,32), and contributes to the development of tolerance. The enzyme responsible for morphine glucuronidation, UGT2B7 (6), is mainly found in the liver, but also exists in the intestines and kidneys (33). Sulfation is a minor pathway (34,35). The metabolites are cleared by the kidneys and partly by biliary excretion. Some recirculation of morphine occurs due to β-glucuronidase activity in the gut (36). Impaired renal function leads to accumulation of M3G and M6G (37).
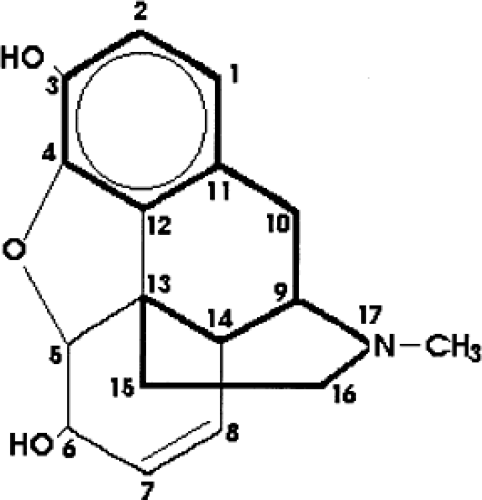 Figure 46.1. The morphine molecule. The structure of morphine and all opium derivatives is characterized by the piperidine ring, which is indicated with bold lines. (From Goodman and Gilman’s The pharmacological basis of therapeutics. New York: Pergamon Press, 1990:485–531, with permission.) |
Morphine may be administered by different routes. Administration in premature newborns is limited to the intravenous route. The painful administration of intramuscular morphine injections is frowned upon. Subcutaneous intermittent boluses through an indwelling catheter offer an alternative route (38). The large variability observed after rectal administration is a major disadvantage of this route, although clinically frequently used (39). Oral morphine, either as elixir or slow-release formulations, offers a good alternative despite a high first-pass effect. Epidural or intrathecal administration may cause delayed respiratory depression due to slow rostral migration within the cerebrospinal fluid (CSF) (40). Patient-controlled analgesia (PCA) is possible in some children aged as young as 6 years, and nurse-controlled analgesia can be used effectively in younger children (41).
Pharmacodynamics
There is a delay between analgesic effect and plasma concentration. The effect compartment equilibration half-time (Teq) for morphine is approximately 17 minutes in adults (42), but is anticipated to be shorted with decreasing age. A concentration–response relationship for morphine analgesia in children has not been described, although adverse effect relationships for both vomiting (43) and respiratory depression are reported (44). The effectiveness of intravenous morphine in infants using validated pain assessment tools has been studied in different age groups. After major surgery, continuous morphine doses of 10 to 40 μg per kg have been shown to be effective in alleviating pain in infants and children 0 to 14 years of age (45). No difference in analgesic effect was found between continuous and intermittent dosing. Randomized controlled trials showed different pharmacodynamic effects of intravenous morphine in premature neonates requiring
artificial ventilation with continuous doses of 10 to 30 μg per kg (46,47). Morphine did not reduce pain responses during endotracheal suctioning (48). Chay et al. reported mean morphine concentrations required to produce adequate sedation in 50% of neonates to be 125 ng per mL (49), but analgesic target plasma concentrations for postoperative morphine analgesia are generally believed to be around 15 to 20 ng per mL (50,51). The large PK and PD variability means that morphine is often titrated to effect using small incremental doses (0.02 mg per kg) in children suffering postoperative pain (52). As morphine should not be routinely prescribed for ventilated newborns, it should be used only in those newborns who have increased pain scores and doses again should be titrated to the individual needs of each neonate (46,47).
artificial ventilation with continuous doses of 10 to 30 μg per kg (46,47). Morphine did not reduce pain responses during endotracheal suctioning (48). Chay et al. reported mean morphine concentrations required to produce adequate sedation in 50% of neonates to be 125 ng per mL (49), but analgesic target plasma concentrations for postoperative morphine analgesia are generally believed to be around 15 to 20 ng per mL (50,51). The large PK and PD variability means that morphine is often titrated to effect using small incremental doses (0.02 mg per kg) in children suffering postoperative pain (52). As morphine should not be routinely prescribed for ventilated newborns, it should be used only in those newborns who have increased pain scores and doses again should be titrated to the individual needs of each neonate (46,47).
Pharmacokinetics
Morphine clearance matures with postconceptual age (6,50) and reaches adult rates at 6 to 12 months (Table 46.3). Fetuses have been shown to be capable of metabolizing morphine from 15 weeks of gestation (53,54). In premature newborns, morphine clearance increases with gestation and postnatal age (48). Morphine plasma concentrations show large between individual variability (55). Clinical circumstances, such as type of surgery, concurrent illness, and infants requiring ECMO (56), also have reduced morphine clearance. Protein binding of morphine is low, from 20% in premature neonates (57) to 35% in adults (58), and has no impact on disposition changes with age (59).
Respiratory depression may occur at concentrations of 20 ng per mL (51). Respiratory depression, as measured by carbon dioxide response curves or by arterial oxygen tension, is similar in children aged 2 to 570 days at the same morphine concentration (44). Furthermore, intrathecal dosing causes similar respiratory depression at similar CSF concentrations in children aged 4 months to 15 years (60). Hypotension, bradycardia, and flushing are part of the histamine response to morphine and are associated with a rapid intravenous bolus administration. Morphine in preterm newborns has only small effects on blood pressure, but the effect might be more significant in extreme premature infants and dependant of the morphine doses (61,62). The incidence of vomiting in children after tonsillectomy is related to morphine dose. Doses above 0.1 mg per kg were associated with a more than 50% incidence of vomiting (43).
Table 46.3 Age-Related Clearances of Morphine and Fentanyl Standardized to a 70-kg Person | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Codeine
Codeine, or methylmorphine, is a morphine-like opioid with 1/10 the potency of morphine. It is mainly metabolized by glucuronidation, but minor pathways are N-demethylation to norcodeine and O-demethylation to morphine. Around 10% of codeine is metabolized to morphine. As codeine’s affinity for opioid receptors is very low, the analgesic effect of codeine is mainly due to its metabolite, morphine (63). The cytochrome P450 (CYP) enzyme CYP2D6 catalyzes the metabolism of codeine to morphine. A genetic polymorphism of this enzyme causes distinct phenotypes responsible for the presence of ultrarapid extensive, extensive, and slow codeine metabolizers in the population (64,65). Between 7% and 10% of the population are believed to be slow metabolizers of codeine (65,66,67), but this percentage has been reported to be much higher (65,66). Although codeine has been shown to cause no analgesic effect in the poor metabolizers, side effects persist (67). High incidences of adverse effects might be expected in patients who have an ultrarapid extensive metabolism. These patients achieve higher morphine concentrations. Codeine metabolites and 10% of unmetabolized codeine are excreted in the urine. The plasma half-life of codeine is 3 to 4 hours.
Codeine can be given by intramuscular, oral, and rectal routes. Intravenous codeine is not recommended because of hypotensive effects (68). Rectal codeine achieves lower concentrations than intramuscular codeine because of incomplete, slower, more variable absorption (69). In children, it is generally given in doses of 1 to 3 mg per kg per day. Codeine is often used in combination with acetaminophen or NSAIDs. The addition of codeine to acetaminophen has been shown to improve postoperative pain relief in infants (70). The analgesic effect of acetaminophen (10 to 15 mg per kg) and codeine (1 to 1.5 mg per kg) was comparable to that of ibuprofen (5 to 10 mg per kg) in children after tonsillectomy (71).
Peak plasma concentration (Cmax) occurs after 1 hour (Tmax) after oral administration. The plasma half-life is 3 to 3.5 hours. The Cmax is reached 30 minutes after intramuscular injection (63). The pharmacokinetics of codeine is poorly described in children despite use over decades. A volume of distribution (V) of 3.6 L per kg and a clearance (CL) of 0.85 L per hour have been described in adults, but there are few data detailing pediatric pharmacokinetic developmental changes. The neonatal half-life is longer due to immature clearance (e.g., 4.5 hours), while that of an infant is shorter (e.g., 2.6 hours) (72). Administration (especially of codeine preparations with an antihistamine and a decongestant) in the neonate may cause intoxication (73). Recently, a newborn died from morphine poisoning when his mother used codeine while breastfeeding. The mother, an ultrarapid metabolizer, produced much more morphine when taking codeine than most people do (74,75). Codeine has been used in infants and neonates
after major surgery as an adjunct to acetaminophen or NSAIDs (optimal oral codeine dosage 1 to 1.5 mg per kg every 4 to 6 hours, oral acetaminophen 20 mg per kg every 6 hours in infants older than 3 months) (76).
after major surgery as an adjunct to acetaminophen or NSAIDs (optimal oral codeine dosage 1 to 1.5 mg per kg every 4 to 6 hours, oral acetaminophen 20 mg per kg every 6 hours in infants older than 3 months) (76).
The adverse effects of codeine are broadly similar to those of other opioids. Adverse effects at low doses appear to be directly related to morphine plasma concentrations, but are caused by codeine at higher doses (77). There is a broad belief that codeine causes fewer side effects, such as sedation and respiratory depression, compared with other opioids, but there is little evidence for this.
The analgesic effect of codeine is dependent on its conversion to morphine. Consequently, other medications competing for the CYP2D6 enzyme (e.g., quinidine) may decrease the analgesic effect of codeine.
Oxycodone
Oxycodone is a semisynthetic analgesic that is available as an immediate-release product (oral solution and capsule) as well as a controlled-release tablet for 12-hourly administration. Immediate-release and controlled-release preparations of oxycodone have similar efficacy and comparable side-effect profiles in adults (78). The relative bioavailability of intranasal, oral, and rectal formulations was approximately 50% that of intravenous in adults. The buccal and sublingual absorption of oxycodone is similar in young children (79). Intramuscular administration provides relatively constant drug absorption, while buccal and gastric administration is associated with large interindividual variation in the rate of absorption (80). Mean values of drug clearance and volume of distribution (Vss) were 15.2 (SD 4.2) mL per minute per kg and 2.1 (SD 0.8) L per kg in children after ophthalmic surgery (81).
Oxycodone is very expensive, and drugs such as controlled-release morphine and methadone offer cheaper alternatives (78). Controlled-release oxycodone may be appropriate if the patient cannot tolerate other controlled-release or long-acting opioid analgesics. Olkkola et al. showed that oxycodone (0.1 mg per kg) in children after ophthalmic surgery caused greater ventilatory depression compared with other opioids (81).
Methadone
Methadone is a synthetic opioid with an analgesic potency similar to that of morphine but with a more rapid distribution and a slower elimination. Methadone is used as a maintenance drug in opioid-addicted adults to prevent withdrawal. Methadone might have beneficial effects because it is a long-acting synthetic opioid with a very high bioavailability by the enteral route. Although only few data on the efficacy and safety of methadone are available, methadone is widely used for the treatment of opioid withdrawal in neonates and children (82,83). Intravenous methadone has been shown to be an effective analgesic for postoperative pain relief (84), and oral administration has been recommended as the first-line opioid for severe and persistent pain in children (85). It also seems to be a safe enteral alternative for intravenous opioids in palliative pediatric oncological patients (86). Although a predominant role for methadone in the management of prolonged pain in neonates has been suggested, use needs to be evaluated in a clinical research setting (87). The few data on methadone pharmacokinetics show a slow elimination half-life with large interindividual variability (3.8 to 62 hours) (87). Methadone’s lipid solubility is greater than that of morphine (88). The increased lipid solubility and longer duration of effect give this drug potential for single-shot epidural use.
Hydromorphone
Hydromorphone is a semisynthetic congener of morphine with a potency of around 5 to 7.5 times that of morphine (89). Hydromorphone is metabolized to hydromorphone-3-glucuronide and also, to a lesser extent, to dihydroisomorphine and dihydromorphine (90). The intravenous:oral dose ratio is 1:5, as there is high first-pass metabolism (91). A clearance of 51.7 mL per minute per kg (range, 28.6 to 98.2) is reported in children (92).
Hydromorphone is used for chronic cancer pain and for postoperative analgesia. The side-effect profile is comparable to that of other strong opioids, and hydromorphone does not convincingly demonstrate clinical superiority in adults over other strong opioid analgesics (93). Goodarzi showed that epidural hydromorphone caused fewer side effects than morphine and fentanyl in children undergoing orthopedic procedures (94). PCA with hydromorphone seems to result in similar analgesia and side effects compared to morphine in children for the management of mucositis pain after bone marrow transplantation (89). Plasma concentrations of around 4.7 ng per mL (range 1.9 to 8.9 ng per mL) relieve mucositis in children given PCA devices. Time to peak concentration is 4 to 6 hours and clearance is 51.7 mL per minute per kg in children (89,92).
Meperidine (Pethidine)
Meperidine is a weak opioid, primarily μ-receptor, agonist that has a potency of 1/10 that of morphine. The analgesic effects are detectable within 5 minutes of intravenous administration and peak effect is reached within 10 minutes (36,95). In adults, meperidine is metabolized to meperidinic acid and normeperidine. Meperidine clearance in infants and children is approximately 10 mL per minute per kg (96,97). Elimination in neonates is greatly reduced, and elimination half-time in neonates, who have received pethidine by placental transfer, may be two to seven times longer than that in adults (98).
Meperidine was initially synthesized as an anticholinergic agent but was soon discovered to have analgesic properties. Although meperidine’s anticholinergic effects were demonstrated in vivo, the anticholinergic effects on the biliary and renal tracts have not been demonstrated in vivo. Studies have clearly demonstrated that meperidine is no more efficacious in treating biliary or renal tract spasm than comparative μ-opioids. Meperidine was portrayed in practice and teaching as having unique clinical advantages (99). Because morphine results in better analgesia with fewer side effects, there are no particular advantages of meperidine as an analgesic (100). Accumulation of the metabolite normeperidine results in seizures and dysphoria (3).
Intramuscular administration of meperidine was frequently used in pediatric patients, but this route of administration is used uncommonly now because it is painful. Meperidine’s local anesthetic properties have been found useful for epidural techniques (101).
Intramuscular administration of meperidine was frequently used in pediatric patients, but this route of administration is used uncommonly now because it is painful. Meperidine’s local anesthetic properties have been found useful for epidural techniques (101).
Fentanyl
Fentanyl is a synthetic opioid that acts as a “morphine-like agonist.” Its potency is about 50- to 100-fold that of morphine, with a large postulated effect on the μ-receptor. A plasma concentration of 15 to 30 ng per mL is required to provide total intravenous anesthesia (102,103). Fentanyl has a wide margin of safety and beneficial effects on hemodynamic stability (104,105) and has a rapid onset (Teq = 6.6 minutes) and a short duration of action. This is probably due to the relative increased lipid solubility and molecular conformation, enabling efficient penetration of the blood–brain barrier. Fentanyl may be the preferred analgesic agent for critically ill patients with hemodynamic instability, patients with symptoms related to histamine release during morphine infusion, or patients with morphine tolerance. Because of its rapid onset of action and short duration of effect, fentanyl efficiently alleviates procedural pain (106). It has been used in neonates on artificial ventilation (107) with bronchopulmonary dysplasia, pulmonary hypertension, and/or diaphragmatic hernia. One study showed a need to escalate dose during ECMO, indicating a rapid development of tolerance (108). Overall, the use of synthetic opioids shows a more rapid tolerance (3 to 5 days) than that of morphine (2 weeks) and heroin (weeks) (87,109).
Fentanyl metabolism is related to the activity of the hepatic cytochrome P450 system (CYP3A4) and is metabolized by oxidative N-dealkylation into norfentanyl and hydroxylized (110,111). All metabolites are inactive, and a small amount of fentanyl is renally eliminated unmetabolized (112).
Fentanyl has been shown to effectively prevent preterm neonates from surgical stress responses and to improve postoperative outcome (113). Single fentanyl doses (3 μg per kg) and infusion (1.1 μg per kg per hour) reduced physiologic and behavioral measures of pain and stress during mechanical ventilation in preterm neonates (114,115) as effectively as morphine (107). International recommended starting doses are, however, smaller, as is shown in Table 46.1. In older infants and children, fentanyl has been shown to be effective for the management of peri- and postoperative pain (116) and for the management of procedural pain. Fentanyl clearance may be impaired due to decreased hepatic blood flow (e.g., from increased intra-abdominal pressure) (117) in neonates after major abdominal surgery (e.g., omphalocele). Fentanyl also has a propensity for muscular rigidity (118). Transdermal fentanyl can be used for severe cancer-related pain (119) or in palliative pediatric care (16). Fentanyl plasma concentrations are not measurable until 2 hours after application, and there is an 8 to 16 hours latency until full clinical fentanyl effects are observed. Following removal, serum fentanyl concentrations decline gradually and fall to 50% in approximately 16 hours. This prolonged apparent elimination half-life occurs because fentanyl continues to be absorbed from the skin, where a fentanyl depot concentrates (120). The systemic availability of fentanyl by this route is approximately 30% of that found using the intravenous route (121). Oral transmucosal fentanyl provides consistent analgesia for brief painful procedures (122). Transdermal and transmucosal fentanyl have not been studied in newborns.
Because fentanyl has very high lipid solubility, it is widely distributed in tissues. Its short duration of effect is due to redistribution to deep, lipid-rich compartments. Accumulation of fentanyl in lipid-rich tissues may redistribute slowly after discontinuation of therapy, resulting in prolonged periods of sedation and respiratory depression after an extended period of use (123). The context-sensitive half-time after an infusion of 1 hour is approximately 20 minutes, but it is 270 minutes after an 8-hour infusion (124).
The clearance of fentanyl appears to be somewhat immature at birth but increases dramatically after birth. Fentanyl clearance is 70% to 80% of adult values in term neonates (Table 46.3) and, standardized to a 70-kg person, appears to reach adult levels within the first 2 weeks of life (123,125). The volume of distribution of fentanyl at steady state is around 5.9 L per kg in term-born neonates and decreases with age to 4.5 L per kg during infancy, 3.1 L per kg during childhood, and 1.6 L per kg in adults (126). Initial plasma concentrations in pediatric patients are lower than in adults due to larger distribution volumes.
The intraoperative use of 3 μg per kg fentanyl in infants did not result in respiratory depression or hypoxemia in a placebo-controlled trial (116). Only 3 of 2,000 nonintubated infants and children experienced short apneic episodes after a low dose of fentanyl for the repair of facial lacerations (127). Fentanyl has similar respiratory depression in infants and adults when plasma concentrations are similar (128).
Fentanyl, alfentanil, and sufentanil are metabolized by CYP3A4, and other drugs that also use this enzyme (e.g., cyclosporine, erythromycin) may decrease clearance, leading to increased fentanyl plasma concentrations (129,130). Acetaminophen has been shown to interact with fentanyl metabolism in vitro (131), although the clinical importance of this interaction is probably unimportant.
Research investigating DNA polymorphisms has shown genetic variability of CYP3A4 with slow and rapid metabolizers. One explanation for between individual variability in clearance across a patient population appears to be the efficiency of drug metabolism arising from differences in enzyme expression levels and/or from the presence of allelic variants of the enzyme with compromised catalytic ability.
Alfentanil
Alfentanil is a synthetic opioid that is chemically a derivate of fentanyl. It has a rapid onset (Teq = 0.9 minute), a brief duration of action, and one-fourth the potency of fentanyl. Alfentanil has lower lipid solubility and causes less histamine release (18) than fentanyl. It is used as a procedural analgesic for pediatric patients because the onset of analgesia is rapid (132). Sufficient analgesia during endotracheal intubation and suctioning has been found using 10 to
20 μg per kg alfentanil in preterm neonates (132,133,134). A target plasma concentration of 400 ng per mL is used in anesthesia. Metabolism is comparable to that in adults, that is, phase 1 via oxidative N-dealkylation by CYP3A4 (99) and O-dealkylation, and then phase 2 conjugation to renally excreted end products (135). Alfentanil plasma protein binding increases from 65% in preterm neonates and 79% in term infants to around 90% in adults (136,137). The volume of distribution is smaller in infants than in adults (138). Clearances of alfentanil, standardized to a 70-kg person, are similar at different ages (250 to 500 mL per minute per 70 kg) except for the neonatal age group, in which clearances are decreased (20 to 60 mL per minute per 70 kg). Consequently, elimination half-life in children (40 to 68 minutes) is higher in the neonatal period. In premature neonates, the half-life is as long as 6 to 9 hours (139,140). Children with chronic renal failure or chronic hepatic disease do not show impaired clearance of alfentanil (141). Alfentanil cannot be used without neuromuscular blocking drugs in newborns because of a very high incidence of rigidity (132,142).
20 μg per kg alfentanil in preterm neonates (132,133,134). A target plasma concentration of 400 ng per mL is used in anesthesia. Metabolism is comparable to that in adults, that is, phase 1 via oxidative N-dealkylation by CYP3A4 (99) and O-dealkylation, and then phase 2 conjugation to renally excreted end products (135). Alfentanil plasma protein binding increases from 65% in preterm neonates and 79% in term infants to around 90% in adults (136,137). The volume of distribution is smaller in infants than in adults (138). Clearances of alfentanil, standardized to a 70-kg person, are similar at different ages (250 to 500 mL per minute per 70 kg) except for the neonatal age group, in which clearances are decreased (20 to 60 mL per minute per 70 kg). Consequently, elimination half-life in children (40 to 68 minutes) is higher in the neonatal period. In premature neonates, the half-life is as long as 6 to 9 hours (139,140). Children with chronic renal failure or chronic hepatic disease do not show impaired clearance of alfentanil (141). Alfentanil cannot be used without neuromuscular blocking drugs in newborns because of a very high incidence of rigidity (132,142).
Sufentanil
Sufentanil is a most potent opioid analgesic, and is 5 to 10 times more potent than fentanyl, with a Teq of 6.2 minutes. A concentration of 5 to 10 ng per mL is required for total intravenous anesthesia, 0.2 to 0.4 ng per mL for analgesia.
Elimination of sufentanil has been suggested by O-demethylation and N-dealkylation in animal studies. Like fentanyl and alfentanil, the P450 CYP3A4 enzyme is responsible for the N-dealkylation (110). The amount of free sufentanil decreases with age (neonates 19%; infants 11%; children/adults 8%) and is strongly correlated with the α-1-acid glycoprotein plasma concentration (136). The lower concentration of α-1-acid glycoprotein in newborns and infants contributes to the increased free fraction of sufentanil in these age groups. Although sufentanil, fentanyl, alfentanil, and remifentanil have high protein binding (>70%) and have high hepatic (or nonhepatic for remifentanil) extraction ratios, protein binding changes are probably clinically unimportant (59) because dose is titrated to effect, and clearance variability has greater impact.
Remifentanil
Remifentanil resembles fentanyl, sufentanil, and alfentanil in chemical structure. It is a selective μ-receptor agonist with a higher potency than alfentanil. Because the inhibitory neurotransmitter glycine is used as a carrier for remifentanil, it should not be used for spinal or epidural applications (143), and because of its short duration of action, it is usually given as an infusion (Teq = 1.16 minutes) (144,145). A concentration of 2 to 4 ng per mL supplements anesthesia for major surgery. Analgesic concentrations are 0.2 to 0.4 ng per mL.
Remifentanil is metabolized to carbonic acid. The metabolism is independent of liver and renal function. Remifentanil reacts with nonspecific esterase in tissue and erythrocytes (146,147). Carbonic acid is excreted through the kidneys. Clearances decreases with age, with rates of 90 mL per kg per minute in infants younger than 2 years, 60 mL per kg per minute between 2 and 12 years of age, and 40 mL per kg per minute in adults (148,149,150). Volume of distribution in children is smaller (200 to 300 mL per kg) than in adults (400 mL per kg), and might by increased in young infants (450 mL per kg). Elimination half-life seems to be constant, around 3 to 6 minutes (40,151).
Administration of 1 μg per kg intravenously followed by 0.1 to 1.0 μg per kg per minute results in sufficient analgesia during surgery in children (152,153,154,155). Because of its short duration, remifentanil seems to be ideally suited for pediatric neurosurgical patients who may require neurologic assessment at completion of surgery (156). Its use is accompanied by a high incidence of life-threatening respiratory depression at subtherapeutic concentrations (157). As a result of a rapid development of μ-receptor tolerance with remifentanil use, higher subsequent opioid doses are required.
Nonopioids
Acetaminophen
Mechanism of Action
Acetaminophen (paracetamol) has been used in clinical practice for more than 100 years and is the most commonly prescribed pediatric medicine. Acetanilid, the parent compound of acetaminophen, was introduced in 1886. Toxicity-related problems with acetanilid led to the introduction of acetaminophen (N-acetyl-p-amino-phenol) by von Mering in 1893. The popularity of acetaminophen over the nonsteroidal anti-inflammatory agents ascended after the reported association between Reye syndrome and aspirin in the 1980s (160). Acetaminophen is widely used in the management of pain and fever but is lacking anti-inflammatory effects.
Acetaminophen has a central analgesic effect that is mediated through activation of descending serotonergic pathways. Debate exists about its primary site of action, which may be inhibition of prostaglandin synthesis or through an active metabolite influencing cannabinoid receptors. Prostaglandin H2 synthetase (PGHS) is the enzyme responsible for metabolism of arachidonic acid to the unstable prostaglandin H2. The two major forms of this enzyme are the constitutive PGHS-1 and the inducible PGHS-2. PGHS comprises two sites: a cyclooxygenase (COX) site and a peroxidise (POX) site. The conversion of arachidonic acid to prostaglandin G2 (PGG2) is dependent on a tyrosine-385 radical at the COX site. Formation of a ferryl protoporphyrin IX radical cation from the reducing agent Fe31 at the POX site is essential for conversion of tyrosine-385 to its radical form. Acetaminophen acts as a reducing cosubstrate on the POX site and lessens availability of the ferryl protoporphyrin IX radical cation. This effect can be reduced by the presence of hydroperoxide-generating lipoxygenase enzymes within the cell (peroxide tone) or by swamping the POX site with substrate such as PGG2. Peroxide tone and swamping explain acetaminophen’s
lack of peripheral analgesic effect, platelet effect, and anti-inflammatory effects. Alternatively, acetaminophen effects may be mediated by an active metabolite (p-aminophenol). P-aminophenol is conjugated with arachidonic acid by fatty acid amide hydrolase to form AM404. AM404 exerts effect through cannabinoid receptors (161).
lack of peripheral analgesic effect, platelet effect, and anti-inflammatory effects. Alternatively, acetaminophen effects may be mediated by an active metabolite (p-aminophenol). P-aminophenol is conjugated with arachidonic acid by fatty acid amide hydrolase to form AM404. AM404 exerts effect through cannabinoid receptors (161).
Pharmacodynamics
Antipyresis
Brown et al. (162) reported a linked-population PK–PD model for antipyresis using mixed-effect models. The acetaminophen PK parameters were similar to those reported by others (163,164). The link component yielded a Keo (ln 2/Teq) of 0.58 (SE 0.06) per hour (Teq 71, SE 7 minutes); the sigmoid Emax component yielded an EC50 of 4.63 g per L and a Hill coefficient N of 3.98 (SE 0.42). The predicted maximum effect response was 1.38#65518;C, but this was possibly constrained by the temperature enrollment criteria and a failure to explore higher doses. These authors noted two major additional factors that influence interpretation of the relationship between plasma concentration and fever reduction: the cyclical nature of fever and the initial temperature at when acetaminophen was given.
Analgesia
Korpela et al. (165) studied day-stay surgical children randomized to receive a single dose of 0, 20, 40, or 60 mg per kg of rectal acetaminophen. In the postanesthesia care unit, pain scores were significantly lower in the 40 and 60 mg per kg groups compared with placebo and 20 mg per kg groups. Acetaminophen resulted in a dose-related reduction in the number of children who required postoperative rescue opioid, with significance reached with 40 or 60 mg per kg doses. Calculated dose of acetaminophen at which 50% of the children did not require a rescue opioid was 35 mg per kg. However, neither the antipyretic nor the analgesic effect of acetaminophen is directly related to plasma concentration. Time delays of approximately 1 hour between peak concentration and peak effect are reported (166,167). The time delay is similar to that described for acetaminophen CSF kinetics (168).
Pain fluctuations, pain type, and placebo effects complicate interpretation of clinical studies (169). Anderson et al. (164) reported pharmacodynamic population parameter estimates [population variability coefficient of variation (CV)] in children after tonsillectomy. An Emax model in which the greatest possible pain relief (visual analogue scale 0 to 10) equates to an Emax of 10 were Emax = 5.17 (64%) and EC50 = 9.98 (107%) mg per L. The equilibration half-time (Teq) of the analgesic effect compartment was 53 (217%) minutes. A target effect compartment concentration of 10 mg per L is associated with a pain reduction of 2.6/10. Effect site concentrations associated with analgesia for the pains experienced as a neonate are unknown.
The use of an intravenous acetaminophen formulation allows greater dosing accuracy, less pharmacokinetic variability attributable to absorption, and more rapid speed of effect onset (169). A new intravenous acetaminophen preparation, with mannitol, cysteine, and sodium phosphate as carriers, has circumvented problems of injection site pain associated with the earlier prodrug (propacetamol). Infusions over 15 minute of intravenous acetaminophen 15 mg per kg following inguinal hernia repair in children resulted in a steep reduction in pain relief between 15 and 30 minute (170), consistent with a delay achieving effect site concentrations in the brain. Intravenous acetaminophen has been successfully used for neonatal analgesia (171) and two European centers have published dosing guidelines in neonates (172,173). These regimens attempt to achieve a steady-state target concentration of 10 mg per L.
Pharmacokinetics
Oral Absorption
Bioavailability. Acetaminophen has low first-pass metabolism, and the hepatic extraction ratio is 0.11 to 0.37 in adults (174). The relative bioavailability of rectal compared with oral acetaminophen formulations (Frectal/oral) has been reported as 0.52 (range 0.24 to 0.98) (175) and even as low as 0.3 (176). The relative bioavailability is higher in neonates, where it is possible suppository insertion height may result in a different rectal venous drainage pattern. Hopkins et al. (177) noted that the area under the curve (AUC) in neonates after suppository administration was significantly greater than in infants and in older children. Van der Marel et al. (178) noted a similar finding in children aged 10.3 (SD 2.3) months after craniofacial surgery. The mean AUC was 171.2 mg per hour per L after rectal suppository compared with 111.9 mg per hour per L after elixir, although some of this difference was attributable to vomiting after oral elixir. The relative bioavailability of rectal formulations appears to be age related. The bioavailability of the capsule suppository relative to elixir decreased with age from 0.92 (22%) at 28 weeks postconception to 0.86 at 2 years of age; the triglyceride base decreased from 0.86 (35%) at 28 weeks postconception to 0.5 at 2 years of age (Fig. 46.2). The relative bioavailability of a rectal solution was 0.66 (179).
Rate of absorption. Acetaminophen has a pKa of 9.5, and in the alkaline medium of the duodenum, acetaminophen is nonionized. Consequently, absorption of the nonionized
form from the duodenum to the systemic circulation is rapid (T1/2, abs = 6.8, SD 0.9 minute in adult volunteers) (180). A pharmacokinetic model using a first-order input model with a lag time has been used to describe absorption. Brown et al. (181) reported rapid absorption (T1/2, abs = 2.7, SE 1.2 minutes, Tlag = 4.2, SE 0.4 minute) parameters in febrile children given elixir orally. Similar absorption half-lives have been estimated in children given acetaminophen as an elixir before tonsillectomy (T1/2, abs = 4.5 minutes, CV = 63%, Tlag = 0) (181). These children had undergone fasting for at least 6 hours prior to surgery, and gastric transit times were rapid. Absorption in children younger than 3 months was delayed by a factor of 3.68 (182). Acetaminophen absorption depends on gastric emptying, and gastric emptying is slow and erratic in the neonate (183). Normal adult rates may not be reached until 6 to 8 months (184). Gastric pH is near neutral in neonates. This increases the unionized form available for absorption for acetaminophen (185). The stomach, however, is a poor route of entry to the circulation. The increased surface area, blood flow, and permeability of the small intestine favor the intestinal route. Oral absorption was considerably delayed in premature neonates in the first few days of life (186).
form from the duodenum to the systemic circulation is rapid (T1/2, abs = 6.8, SD 0.9 minute in adult volunteers) (180). A pharmacokinetic model using a first-order input model with a lag time has been used to describe absorption. Brown et al. (181) reported rapid absorption (T1/2, abs = 2.7, SE 1.2 minutes, Tlag = 4.2, SE 0.4 minute) parameters in febrile children given elixir orally. Similar absorption half-lives have been estimated in children given acetaminophen as an elixir before tonsillectomy (T1/2, abs = 4.5 minutes, CV = 63%, Tlag = 0) (181). These children had undergone fasting for at least 6 hours prior to surgery, and gastric transit times were rapid. Absorption in children younger than 3 months was delayed by a factor of 3.68 (182). Acetaminophen absorption depends on gastric emptying, and gastric emptying is slow and erratic in the neonate (183). Normal adult rates may not be reached until 6 to 8 months (184). Gastric pH is near neutral in neonates. This increases the unionized form available for absorption for acetaminophen (185). The stomach, however, is a poor route of entry to the circulation. The increased surface area, blood flow, and permeability of the small intestine favor the intestinal route. Oral absorption was considerably delayed in premature neonates in the first few days of life (186).
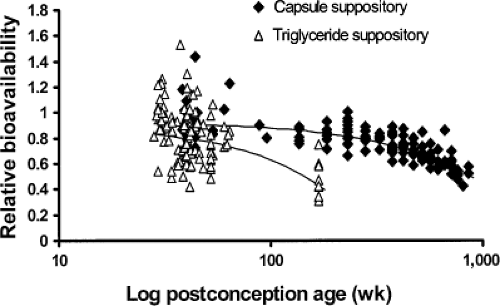 Figure 46.2. Changes in relative bioavailability of triglyceride-based and capsule suppository formulations with age. (From Anderson BJ, van Lingen RA, Hansen TG, et al. Acetaminophen developmental pharmacokinetics in premature neonates and infants: a pooled population analysis. Anaesthesiology 2002;96: 1336–1345, with permission.) |
Rectal Absorption
Rectal acetaminophen is widely used in children presenting for surgical procedures because of gastrointestinal dysfunction or preoperative fasting guidelines that disallow food or fluids up to 6 hours before surgery. It is also preferable in children with nausea and vomiting. Some cultures have a greater preference for this route over oral administration (e.g., France vs. England (187). Absorption through the rectal route is slow and erratic (188,189). Data in preterm neonates are scarce (190).
Birmingham et al. (191) reported a first-order input model with a zero-order dissolution time to describe absorption characteristics of a triglyceride-base suppository in children. Others have reported a first-order absorption model with lag time. Regardless of the model, absorption is slow, with large variability (192). For example, absorption parameters for the triglyceride base and capsule suppository were T1/2, abs = 1.34 hours (CV = 90%), Tlag = 0.14 hour (31%) and T1/2, abs = 0.65 hour (63%), Tlag = 0.54 hour (31%). The absorption half-life for rectal formulations was prolonged in infants younger than 3 months (1.51 times greater) compared with values in older children (168).
Clearance
Several studies have shown that the rate constant for acetaminophen glucuronide formation in neonates is considerably smaller than in adults, but the rate constant for sulfate formation is larger than in adults (190,193,194). Glucuronide/sulfate ratios range from 0.12 in premature neonates of 28 to 32 weeks postconception age to 0.28 in those at 32 to 36 weeks postconception to 0.34 in term neonates 0 to 2 days old. Ratios of 0.75 in children aged 1 to 9 years, 1.61 in those aged 12 years, and 1.8 in adults were reported. Approximately 4% of acetaminophen is excreted in urine unmetabolized and the amount is dependent on urine flow (195).
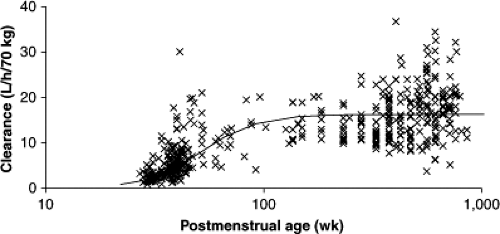 Figure 46.3. Clearance changes with age. Individual predicted clearances, standardized to a 70-kg person, from the NONMEM program’s post hoc step are plotted against age. The thin line demonstrates the predicted nonlinear relationship between clearance and age. (From Anderson BJ, Allegaert K. Intravenous neonatal acetaminophen dosing; the magic of ten days. Pediatr Anesth 2009;19:289–295, with permission.) |
Population parameter estimates (between subject variability, %) were central volume (V2/Foral) 24.0 (54.7%), peripheral volume of distribution (V3/Foral) 30.4 (31.7%), clearance (CL/Foral) 16.3 (40.4%), and intercompartment clearance (Q/Foral) 54.7 (116%). Clearance increased from 27 weeks PCA (1.87 L per hour per 70 kg) to reach 84% of the mature value by 1 year of age (standardized to a 70-kg person using allometric “¼ power” models) (196). Figure 46.3 shows age-related clearance estimates. Between occasion variability for clearance (CL/Foral) was 18.5%.
Volume of Distribution
The population distribution volumes are similar to those reported in adults (56 to 70 L) (197). The volume of distribution for acetaminophen in mammals (197), including humans, is 49 to 70 L per 70 kg, as we would expect from the allometric size model with a power function of 1. Peripheral volume of distribution decreased from 27 weeks PCA (45.01 per 70 kg) to reach 110% of its mature value by 6 months of age (Fig. 46.4). Central volume of distribution and intercompartment clearance did not change with age. Between occasion variability for the peripheral volume of distribution (V3/Foral) was 19.3% (196).
Fetal body composition and water distribution alter considerably during the third trimester, and over the first few months of life probably reflects neonatal body composition and the rapid changes in body water distribution in early life because acetaminophen has a low molecular weight and reasonable lipid solubility (198) and acetaminophen crosses cell membranes easily, distributing throughout all tissues and fluids (199). The tissue:plasma concentration ratios are close to unity at equilibrium (199).
Disease States
The effects of altered physiology such as fever (200), anesthesia (201), or mildly impaired hepatic dysfunction (202,203) on pharmacokinetic parameters have received little attention, but appear to have minimal impact in children.
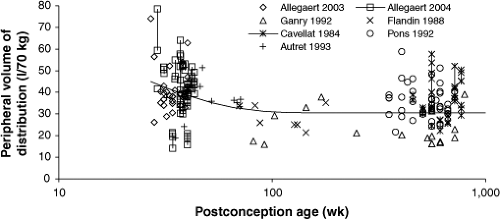 Figure 46.4. Peripheral volume of distribution changes with age. Individual predicted volumes, standardized to a 70-kg person, against age. The solid line demonstrates the nonlinear relationship between volume of distribution and age. Those children given multiple doses have volume of distribution estimates linked by a fine line to demonstrate between occasion variability. (From Anderson BJ, Pons G, Autret-Leca E, et al. Paediatric intravenous acetaminophen (propacetamol) pharmacokinetics; a population analysis. Pediatr Anesth 2005;15:282–292, with permission.) |
Toxicity
Chronic Use of Acetaminophen
Acetaminophen overdose results in increased production of highly reactive electrophilic arylating metabolites by the hepatic cytochrome P450-dependent mixed-function oxidase enzyme system (204). The toxic metabolite of acetaminophen, N-acetyl-p-benzoquinone imine (NAPQI), is formed by the CYP2E1, 1A2, and 3A4 (205). This metabolite binds to intracellular hepatic macromolecules to produce cell necrosis and damage. Acetaminophen concentrations may rise in pediatric patients with low clearance after regular doses of 15 mg per kg every 4 hours (206). There is evidence of glutathione depletion in adult volunteers given doses of 0.5 and 3 g acetaminophen separated by 4 to 10 days (207). Penna and Buchanan (208) reported 7 deaths and 11 cases of hepatotoxicity associated with acetaminophen in children. Mortality due to hepatotoxicity was associated with doses greater than 300 mg per day per kg for 1 to 6 days. Survival was usually seen in those children suffering hepatotoxicity due to acetaminophen greater than 150 mg per day per kg for 2 to 8 days. Subsequent guidelines (209,210) recommend that doses should not exceed 90 mg per day per kg.
Significant hepatic and renal disease, malnutrition, and dehydration increase the propensity for toxicity. Medications that induce the hepatic CYP2E1, 1A2, and 3A4 systems (e.g., phenobarbitone, phenytoin, and rifampicin) may also increase the risk of hepatotoxicity. The coingestion of therapeutic drugs, foodstuffs, or other xenobiotics has potential to induce these enzymes (211). The influence of disease on acetaminophen toxicity is unknown. It has been speculated that ingestion of acetaminophen increases the potential for liver injury by another cause, such as a viral agent (212). Hepatotoxicity causing death or requiring liver transplantation has been reported with doses above 75 mg per kg per day in children and 90 mg per kg per day in infants (211,213,214,215). It is possible that even these regimens may cause hepatotoxicity if used for longer than 2 to 3 days (211). These reports (211,213,214,215) and others from Australia (216,217) and Scotland (218) are concerning. Kozer et al. (219) demonstrated that ill children receiving repeated large doses of acetaminophen (>90 mg per kg per day) may show abnormalities in liver function. It is unknown whether there is a difference in the propensity to toxicity between children given acetaminophen for fever and those given acetaminophen for postoperative analgesia.
Hepatic enzyme profiles have been used as a surrogate assessment in two neonatal studies (220,221). Neither group of authors noted hepatic changes during the treatment periods that extended over a median of 4 days, but the value of hepatic changes is debated (222). It is currently impossible to predict which individuals have an enhanced susceptibility to cellular injury from acetaminophen. One area of future investigation is the role of single nuclear polymorphisms in NAPQI production.
Single Dose of Acetaminophen
The plasma concentration associated with toxicity after a single dose of acetaminophen in children is extrapolated from adult data. The Rumack and Matthew (223) acetaminophen toxicity nomogram is widely used to guide management of acetaminophen overdose in adults and children. This nomogram was derived from a study by Prescott et al. (224) of 30 adult patients who ingested an overdose of acetaminophen. In poisoned patients with similar initial concentrations, the half-life in those without liver damage was 2.9 (SE 0.3) hours and in those with liver damage was 7.6 (SE 0.8) hours. Acetaminophen concentrations of more than 300 mg per L at 4 hours were always associated with severe hepatic lesions, but none were observed in patients with concentrations less than 150 mg per L. The half-life was less than 4 hours in all patients without liver damage.
Clearance is a nonlinear function of weight, whereas volume is a linear function of weight. Dose is usually expressed as a linear function of weight. The 4-hour concentration in children is determined by clearance, not volume, because absorption is rapid after oral elixir. As a consequence, younger children (1 to 5 years) require larger doses than older children and adults to achieve similar concentrations at 4 hours (225,226). This has been demonstrated in animals. Young rats have a higher median lethal dose than older rats (227). More drug is required to produce a hepatotoxic reaction (227). Children (1 to 5 years) with reported accidental ingestion of more than 250 mg per kg (compare to 150 mg per kg in adults) can have serum concentration measured at 2 hours after ingestion rather than the 4-hour time point recommended in adults.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


